This is your very first post. Click the Edit link to modify or delete it, or start a new post. If you like, use this post to tell readers why you started this blog and what you plan to do with it.

This is the post excerpt.
This is your very first post. Click the Edit link to modify or delete it, or start a new post. If you like, use this post to tell readers why you started this blog and what you plan to do with it.
CLIMA LABORAL
El clima laboral no es otra cosa el medio en el que se desarrolla el trabajo cotidiano. La calidad de este clima influye directamente en la satisfacción de los trabajadores y por lo tanto en la productividad empresarial. Si eres capaz de conseguir una mayor productividad con un buen clima laboral, tienes todo lo necesario para conseguir grande éxitos en tu empresa.
De aquella manera, mientras que un buen clima se orienta hacia los objetivos generales, un mal clima destruye el ambiente de trabajo ocasionando situaciones de conflicto, malestar y generando un bajo rendimiento.
los objetivos generales, un mal clima destruye el ambiente de trabajo ocasionando situaciones de conflicto, malestar y generando un bajo rendimiento.
La calidad del clima laboral se encuentra íntimamente relacionado con el manejo social de los directivos y las ventajas y desventajas del liderazgo empresarial, con los comportamientos de los trabajadores, con su manera de trabajar y de relacionarse, con su interacción con la empresa, con las máquinas que se utilizan y con las características de la propia actividad de cada uno.
Propiciar un buen clima laboral es responsabilidad de la alta dirección, que con su cultura y con sus sistemas de gestión, prepararán el terreno adecuado para que se desarrolle. La organización de la empresa deberá estar pensada, entre otras cuestiones, para generar un buen ambiente de trabajo para todos los empleados.
Las políticas de personal y de recursos humanos la mejora de ese ambiente con el uso de técnicas precisas como escalas de evaluación para medir el clima laboral.
El clima laboral u organizacional es un fenómeno complejo, dinámico y multidimensional que presenta las siguientes variables:
El tema del clima laboral ha sido investigado de manera bastante exhaustiva y profunda en las últimas décadas, de ahí que se hayan identificado plenamente los siguientes factores que influyen directamente en la calidad del clima laboral:
Liderazgo. Este factor se refiere al tipo de relación que existe entre jefes y subordinados y el impacto de la misma en el ambiente laboral, y por lo tanto, en la productividad de la empresa. Dentro de los muchos enfoques que la teoría administrativa ha desarrollado al respecto, se sabe que lo mejor es contar con un liderazgo flexible y adaptable. Es decir, el líder deberá tener una amplia gama de actitudes ante las diferentes circunstancias; a veces se deberá ser fuerte, a veces comprensivo.
Relaciones interpersonales. El tipo de relaciones que se crean entre el personal deben ser sanas y fluidas, pues esto afecta a su vez el ánimo de la empresa en general. Es necesario vigilar las relaciones, y estar atento a disgustos y malentendidos entre el personal.
Implicación. Se refiere al grado de compromiso que sienten los empleados hacia la empresa y que en muchas ocasiones está determinado por la percepción del compromiso que la empresa tiene para con sus empleados. Los empleados muestran mayor compromiso en las empresas que tienen la mejor calidad, las mejores ventas y la mejor productividad.
Organización. Se refiere a los elementos que le dan estructura a la empresa, por ejemplo: los puestos, las políticas, los procedimientos, los manuales de operación, etcétera. En el caso de las PYMES, muchas veces la estructura de la empresa está poco definida, y el propietario desempeña un sin número de actividades, desde las operativas hasta las directivas. Por lo tanto, al ir creciendo deberá tener claro que actividades seguirá realizando y cuáles delegará.
Reconocimiento. Se suele decir que cuando alguien hace algo bueno nadie lo recuerda, pero hay un error, todos te lo recuerdan. El reconocer el trabajo bien realizado es vital para contribuir a la formación de un buen ambiente laboral. La psicología organizacional ha comprobado que cuando una persona cree que es buena en alguna actividad, disfrutará al realizarla y lo hará cada vez mejor, lo que impactará su productividad. No desaproveche la oportunidad de reconocer al personal por cada trabajo bien realizado.
Incentivos. Se observa que las empresas que tienen esquemas de remuneración poco dinámicos son las que presentan mayor rotación entre su personal, pues al ganar siempre lo mismo se refuerza la actitud de que no importa el esfuerzo porque siempre se ganará lo mismo. En la actualidad muchas compañías están optando por esquemas compensación dinámica en donde se premie de alguna forma el esfuerzo. Podría creerse que esto solo puede aplicarse a los departamentos de ventas, sin embargo puede ser aplicado a cualquier departamento o empresa, pues cada uno debe tener sus metas y objetivos y en base a esto se puede crear un esquema que fomente en los empleados el deseo por esforzarse mas.
Igualdad. Aunque no todas las personas reaccionan de la misma manera a los mismos estímulos, es necesario dar el mismo trato a todo mundo. Hay que buscar otorgar las mismas condiciones y oportunidades a todos los empleados. Trata de evitar el favoritismo, ya que este fomenta envidias entre el personal y la discordia nunca es sana para el clima laboral. El buen líder conoce a su personal y sabe como motivarlo, reconociendo a las piezas débiles y a los pilares del grupo.
AMBIENTE ÉTICO
El ambiente ético son aquellas ideas que modulan y determinan nuestro andar en un mundo compartido con otros, especialmente que transforman nuestra relación con otros a partir de la pregunta ¿cómo debemos vivir? A veces esta pregunta se responde con un clamor mesiánico, esperando ser guiados por un ser superior o con ínfulas de ser superior que conoce qué es mejor para todos. Ese clima ético llevó a los totalitarismos del siglo XX, cuyo trágico desenlace es bien conocido. A veces esa pregunta se responde por una primacía absoluta del lenguaje de los “derechos”, en el que el marco jurídico es lo único que debe guiar nuestra relación con otros y aquello que la ley no me prohíbe está permitido como parámetro de mi acción. Este clima ético es importante, pero limita las posibilidades de una comunidad a un marco jurídico que, en ocasiones, llega tarde y en otras llega incompleto, permitiendo a unas comunidades la protección jurídica y a otras las deja en vilo. Y a veces la pregunta es respondida con una convicción dogmática e ingenua que solo un grupo quiere el bien para la comunidad y los otros apenas son enemigos de ella. Esta última idea ha permeado el clima ético actual en Colombia que sufre una polarización espeluznante de la que cada vez es más difícil de salir y que presenta un falso dilema: “o estás de acuerdo con nuestros valores o eres un enemigo para la existencia de la comunidad”.
La eutanasia es la intervención voluntaria que acelera la muerte de un paciente desahuciado, con su consentimiento, con la intención de evitar sufrimiento y dolor. La eutanasia está asociada al final de la vida sin sufrimiento.
En un sentido más contemporáneo y restringido, la eutanasia es aquel procedimiento voluntario, intencionado, estudiado y consciente que realiza un médico para acelerar la muerte de un paciente terminal de algún padecimiento incurable; a solicitud consciente, estudiada y deliberada del enfermo o familiares, quienes, plenamente enterados de que no existe tratamiento curativo para la dolencia; le solicitan al médico que la realice sobre el paciente para así dar fin con el dolor y sufrimiento intolerables e intratables.
Existen diferentes leyes sobre la eutanasia en cada país. El Comité selecto de Ética médica de la Cámara de los Lores británica define la eutanasia como una intervención deliberada emprendida con la intención expresa de poner fin a una vida, para aliviar el sufrimiento intratable. En los Países Bajos y en Bélgica, es entendida como «la terminación de la vida por un médico a petición de un paciente. Sin embargo, la ley holandesa no usa el término eutanasia, sino que lo incluye bajo la definición más amplia de suicidio asistido y finalización de la vida a petición. En Colombia la Corte Constitucional en su sentencia C 239 de 1997 manifiesta que el homicidio por piedad es es la acción de quien obra por la motivación específica de poner fin a los intensos sufrimientos de otro, y que doctrinariamente se le ha denominado homicidio pietístico o eutanásico. No obstante, en la sentencia T 970 de 2014, se lee que las definiciones sobre eutanasia son múltiples y actualmente no se cuenta con alguna totalmente aceptada pero aún así, se utiliza la definición de la doctrina jurídica para precisar los elementos necesarios para que el homicidio corresponda al concepto doctrinario de eutanasia; también usa la descripción doctrinaria para su clasificación.
La eutanasia está clasificada de diferentes formas: directa e indirecta según el accionar médico, y voluntaria e involuntaria si se cuenta o no con el consentimiento del paciente.
La eutanasia se practicó en las antiguas Grecia y Roma. Por ejemplo, la cicuta se empleó en la isla de Ceos como un medio para acelerar la muerte; técnica que también se empleaba en Marsella. La eutanasia, en el sentido de la deliberada aceleración de la muerte de una persona, fue apoyada por Sócrates, Platón y Séneca el Viejo en el mundo antiguo, aunque parece que Hipócrates había hablado en contra de la práctica, cuando escribió: no prescribiré una droga mortal para complacer a alguien, ni dar consejos que puedan causar su muerte, lo que indica que pudo haber un cierto debate en la literatura sobre si se pretendía o no incluir la eutanasia.
El concepto de eutanasia en el sentido de aliviar el proceso de la muerte se remonta al historiador médico, Karl Friedrich Heinrich Marx (1796-1877) quien se basó en las ideas filosóficas de Bacon. Según Marx, un médico tenía el deber moral de aliviar el sufrimiento de la muerte mediante el aliento, el apoyo y la mitigación mediante el uso de medicamentos. Tal alivio de la muerte reflejó el espíritu de la época de la cual fue contemporáneo, pero Marx lo colocó en el canon de la responsabilidad médica por primera vez. También hizo hincapié en la distinción entre el cuidado teológico del alma de las personas enfermas desde el cuidado físico y el tratamiento médico por parte de los galenos.
La eutanasia, en su sentido moderno, ha sido fuertemente opuesta a la tradición judeocristiana. Tomás de Aquino (1225-1274) se opuso, y argumentó que la práctica de la eutanasia contradecía nuestros instintos humanos naturales de supervivencia, así como también lo hicieron François Ranchin (1565-1641), médico francés y profesor de medicina y Michael Boudewijns (1601-1681), médico y profesor. Otras voces abogaron por la eutanasia, como el poeta inglés John Donne (1572-1631) en 1624, y la eutanasia continuó en práctica. En 1678, la publicación del libro De pulvinari morientibus non-subtrahend (del latín La almohada de los moribundos no debe ser sustraída) de Caspar Questel, debate sobre el tema. Questel describió varias costumbres que eran usadas en ese momento para traer la muerte a los moribundos, incluida el retiro de la almohada que, se creía, aceleraba la muerte; argumentó en contra de tal práctica, pues hacerlo está contra las leyes de Dios y de natura. Este punto de vista fue compartido por otros que les siguieron, inlcuidos Philipp Jakob Spener, Veit Riedlin y Johann Georg Krünitz. A pesar de la oposición, la práctica de la eutanasia continuó, involucrando técnicas como la sangría, la asfixia y sacar a las personas de sus camas para colocarlas en el suelo frío.
A mediados del siglo xix, surgió el uso de la morfina para tratar «los dolores de la muerte». En 1848 el cirujano estadounidense John Collins Warren (1778-1856) recomendó su empleo. En 1866, el médico británico Joseph Bullar (1815-¿?) reveló una utilización similar para el cloroformo. Sin embargo ninguno de los dos recomendaba que la ocupación de este fármaco debería ser para acelerar la muerte. En 1870, el inglés y maestro de escuela Samuel Williams, inició el debate sobre la eutanasia contemporánea a través de un discurso en el Birmingham Speculative Club, una sociedad cuyos miembros eran filósofos aficionados que recopilaba sus trabajos.
El ensayo fue revisado favorablemente en el diario The Saturday Review de Londres; pero apareció una editorial contra el ensayo en la revista semanal británica The Spectator. A partir de ese momento, resultó ser influyente, y otros escritrores se manifestaron a favor de tales puntos de vista: Lionel Tollemache, octavo conde de Dysart (1794-1878) escribió a favor de la eutanasia, al igual que la británica Annie Besant (1847-1933), la ensayista y reformadora que más tarde se involucró con la National Secular Society (Sociedad Nacional Laica), considerando que era un deber con la sociedad que uno debe morir voluntariamente y sin dolor cuando uno llega al punto de convertirse en una carga. La revista Popular Science analizó el tema en mayo de 1873, evaluando ambos lados del argumento.
El auge del movimiento de la eutanasia en los Estados Unidos de América coincidió con la llamada Edad chapada en oro de ese país, un momento de cambio social y tecnológico que abarcaba un conservadurismo individualista que elogiaba la doctrina económica del laissez faire (en francés: dejen hacer), el método científico y el racionalismo, que sucedió junto a grandes depresiones económicas, industrialización y conflicto entre corporaciones y sindicatos. También fue el período en el que se desarrolló el sistema hospitalario moderno, que ha sido visto como un factor en el surgimiento del debate sobre la eutanasia.
El abogado Robert G. Ingersoll (1833-1899) intercedió a favor de la eutanasia, afirmando, en 1894, que cuando alguien padece una enfermedad terminal, como un cáncer en fase terminal, debería tener derecho a finalizar con su dolor mediante el suicidio. El judío, racionalista intelectual Felix Adler (1851-1933) ofreció un enfoque similiar, aunque, a diferencia de Ingersoll, Adler no rechazó a la religión. De hecho, argumentó un marco de cultura ética. Este último argumentó en 1891 que aquellos que sufrían de un dolor abrumador deberían tener el derecho a suicidarse y, además, que un médico debería estar autorizado para ayudarle. Así Adler, se convierte en el primer estadounidense «prominente» en abogar por el suicidio en casos donde la gente sufría una enfermedad crónica. Tanto Ingersoll como Adler argumentaron a favor de la eutanasia voluntaria en adultos que padecen dolencias terminales. Dowbiggin sostiene que al romper las objeciones morales previas a la eutanasia y el suicidio, Ingersoll y Adler permitieron a otros extender la definición de eutanasia.
El primer intento en este país para legalizar la eutanasia tuvo lugar cuando Henry Thomas Hunt lo introdujo en la Asamblea General de Ohio de 1906. Esto lo hizo Hunt a costa de Anna S. Hall una rica heredera que fue una figura importante en el movimiento de la eutanasia durante los primeros años del siglo xx en los Estados Unidos de América. Hall había visto morir a su madre después de una larga batalla contra un cáncer hepático y se había dedicado a garantizar que los demás no tuvieran que soportar el mismo sufrimiento. Con este fin, participó en una extensa campaña de redacción de cartas, reclutó a Lurana W. Sheldon y a Maud Ballington Booth, y organizó un debate sobre la eutanasia en la reunión anual de la American Humane Association (Asociación Humana Estadounidense) en 1905, descrita por Jacop Appel como el primer debate público significativo sobre el tema en el siglo xx.
El proyecto de ley de Hunt requería la administración de un anestésico para provocar la muerte de un paciente, siempre y cuando la persona sea mayor de edad y tenga la mente sana, y se encuentre sufriendo de una lesión fatal o una enfermedad irrevocable o un gran dolor físico. También requería que el caso fuese atendido por un médico, el consentimiento informado ante tres testigos y la asistencia de tres médicos que tenían que aceptar que la recuperación del paciente era imposible. Una moción para impugnar el proyecto fue rechazada, pero, de todas formas, el proyecto de ley no pasó pues obtuvo una votación de 79 en contra y 23 a favor.
Junto con la proposición de eutanasia del estado de Ohio, en 1906 el asambleísta Ross Gregory presentó una propuesta para permitir la eutanasia a la legislatura de Iowa. Sin embargo, la legislación de Iowa tenía un alcance más amplio que el ofrecido en Ohio. Permitió la muerte de cualquier persona de al menos diez años de edad que sufriere una dolencia que resultaría fatal y causaría un dolor extremo, en caso de que tuvieran una mente sana y expresasen el deseo de apresurar artificialmente su muerte. Además, permitía que los bebés fuesen sacrificados si estaban lo suficientemente deformados, y les permitía a los tutores solicitar la eutanasia en nombre de sus pupilos. La legislación también impuso sanciones a los médicos que se negaren a realizar la eutanasia cuando les fuere solicitada: una pena en prisión de entre seis a doce meses y el pago de una multa entre 200 a 1000 dólares estadounidenses. La propuesta resultó ser controversial; engendrando un debate considerable y no fue aprobada al haberse retirado la consideración después de pasarla a la Comisión de Salud Pública.
Después de 1906, el debate sobre la eutanasia se redujo en intensidad, resurgiendo periódicamente, pero no volviendo al mismo nivel de discusión hasta la década de 1930 en el Reino Unido.
El oponente a la eutanasia, Ian Dowbiggin (1952) argumenta que la creación temprana de la Sociedad Estadounidense pro Eutanasia (ESA; por sus siglas en inglés) reflejó la cantidad de procedimientos eutanásicos percibidos en ese momento, 1920, a menudo viéndolo como un asunto de eugenesia más que como un tema relacionado con los derechos individuales. Dowbiggin sostiene que no todos los eugenistas se unieron a la ESA «solo por razones eugenésicas», si no que, según postula, había claras conexiones ideológicas entre los movimientos eugenésicos y la eutanasia.
La Sociedad Voluntaria de Legalización de la Eutanasia (actualmente denominada Dignity in Diying), fue fundada en 1935 por Charles Killick Millard. El moviento hizo campaña para la legalización de la eutanasia en Gran Bretaña.
En enero de 1936, el rey Jorge V recibió una dosis fatal de morfina y cocaína para acelerar su muerte. En ese momento padecía de insuficiencia cardiorrespiratoria y la decisión de dar fin a su vida la tomó su médico lord Bertrand Dawson. Aunque este evento fue mantenido en secreto durante más de cincuenta años, la muerte de Jorge V coincidió con la legislación propuesta en la Cámara de los Lores para legalizar la eutanasia.
El Aktion T4 es el nombre que se le dio, en la posguerra, al asesinato en masa mediante la eutanasia involuntaria durante la Alemania nazi. La partícula T4 es una abreviación de Tiergartenstraße 4, que era la dirección del departamento de la Cancillería, creado en la primavera de 1940, en el barrio berlinés de Tiergarten, institución que reclutó y pagó al personal asociado con el T4. Ciertos médicos alemanes fueron autorizados a seleccionar pacientes «considerados incurablemente enfermos, después del examen médico más crítico» y luego administrarles una «muerte por piedad» (Gnadentod). Después del final nominal del programa, los médicos en instalaciones alemanas y austríacas continuaron con muchas de las prácticas del Aktion T4, hasta la derrota de la Alemania en 1945.
Los asesinatos tuvieron lugar desde septiembre de 1939 hasta el final de la guerra Mundial en 1945, tiempo durante el cual fueron liquidadas entre 275 000 a 300 000 personas en varios centros de exterminio ubicados en hospitales psiquiátricos en Alemania y Austria, junto con los de la Polonia dominada, y los del Protectorado de Bohemia y Moravia (ahora República Checa). El número de víctimas registradas inicialmente fue un desalentador total de 70 273 personas; el cual ha sido revisado, mostrándose notoriamente al alza, debido al descubrimiento de víctimas adicionales que figuran en los archivos de la antigua Alemania Oriental. Aproximadamente la mitad de los asesinados fueron tomados de los asilos de las iglesias, a menudo con la aprobación de las autoridades protestantes o católicas de esas instituciones.
A pesar de que la Santa Sede anunció el 2 de diciembre de 1940 que la política era contraria a la ley divina natural y positiva y que «el asesinato directo de una persona inocente no esta permitido, ya sea por defectos mentales o físicos», la declaración no fue confirmada por algunas autoridades católicas en Alemania. Por otro lado, durante el verano de 1941, las protestas fueron dirigidas en ese país por el obispo von Galen, cuya intervención, según Richard J. Evans, condujo al «movimiento de protesta más fuerte, explícito y extendido contra cualquier política desde el comienzo del Tercer Reich».
Han sido ofrecidas varias razones para el programa, incluida la eugenesia, la compasión, la reducción del sufrimiento, la higiene racial, la rentabilidad y la presión sobre el presupuesto de beneficencia social. La continuación no oficial de la política dio lugar a muertes adicionales por medicamentos y medios similares, lo que resultó en 93 521 camas vaciadas a finales de 1941. La tecnología que fue desarrollada bajo el programa Aktion T4, particularmente el uso del gas letal para matar a un gran número de personas, fue responsabilidad de la división médica del Ministerio del Interior del Reich, junto con el personal que había participado en el desarrollo de la misma y luego participó en la Operación Reinhard.
La tecnología, el personal y las técnicas desarrollas fueron fundamentales para la implementación de los genocidios nazis. Aunque el programa fue autorizado por Hitler, los homicidios han sido vistos como asesinatos en Alemania. El número de muertos fue aproximadamente unos 200 000 en Alemania y Austria; en otros países europeos, aproximadamente 100 000 personas también fueron víctimas letales.
En el entendimiento actual, el uso del término «eutanasia» en el contexto del Aktion T4 se le considera un eufemismo para ocultar un programa de genocidio, en el cual las personas fueron asesinadas por discapacidades, creencias religiosas y valores individuales discordantes con el régimen nazi. Comparado con las discusiones sobre eutanasia que siguieron al finalizar la guerra, el programa Nazi pudo haber sido redactado en palabras que parecen similares al uso moderno del término, la diferencia radica en que durante el T4 no hubo misericordia y los pacientes no fueron necesariamente pacientes terminales. A pesar de estas diferencias, el historiador y opositor a la eutanasia Ian Dowbiggin escribe que los orígenes de la eutanasia Nazi, como los del movimiento estadounidense pro eutanasia, preceden al Tercer Reich y se entrelazaron con la historia de la eugenesia y el darwinismo social, como también con los esfuerzos para desacreditar la moralidad tradicional y la ética.
El 6 de enero de 1949, la Sociedad Estadounidense pro Eutanasia presentó a la Legislatura del Estado de Nueva York una petición para legalizar la eutanasia, firmada por 379 ministros protestantes y judíos, el grupo más grande de líderes religiosos que haya adoptado esta postura. Una petición similar había sido enviada a la Legislatura de Nueva York en 1947, firmada por aproximadamente mil médicos de Nueva York. Los líderes religiosos católicos criticaron la petición, diciendo que tal proyecto de ley legalizaría un pacto “asesinatosuicida”» dicha crítica incluía una «racionalización del quinto mandamiento de la ley de Dios: “No matarás”.
La petición provocó tensiones entre la Sociedad Estadounidense pro Eutanasia y la Iglesia Católica, lo que contribuyó a un clima de sentimiento anticatólico en general, en relación con cuestiones como el control de la natalidad, la eugenesia y el control de la población. Sin embargo, la petición no dio lugar a ningún cambio legal.

articulo: art13
La bioética es la rama de la ética dedicada a proveer los principios para la conducta más apropiada del ser humano con respecto a la vida, tanto de la vida humana como del resto de seres vivos, así como al ambiente en el que pueden darse condiciones aceptables para la misma.
Se trata de una disciplina relativamente nueva, y el origen del término corresponde al pastor protestante, teólogo, filósofo y educador alemán Fritz Jahr, quien en 1927 usó el término Bio-Ethik en un artículo sobre la relación ética del ser humano con las plantas y los animales. Más adelante, en 1970, el bioquímico estadounidense dedicado a la oncología Van Rensselaer Potter utilizó el término bio-ethics en un artículo sobre la ciencia de la supervivencia y posteriormente en 1971 en su libro Bioética un puente hacia el futuro.
En su sentido más amplio, la bioética, a diferencia de la ética médica, no se limita al ámbito médico, sino que incluye todos los problemas éticos que tienen que ver con la vida en general, extendiendo de esta manera su campo a cuestiones relacionadas con el medio ambiente y al trato debido a los animales. Se han formulado una serie de definiciones respecto a la disciplina de la Bioética, siendo una de ellas la adoptada por la Unidad Regional de Bioética de la OPS, con sede en Santiago de Chile y que, modificada por el S. J. Alfonso Llano Escobar en una revista de la especialidad, define a la Bioética como el uso creativo del diálogo inter y transdisciplinar entre ciencias de la vida y valores humanos para formular, articular y, en la medida de lo posible, resolver algunos de los problemas planteados por la investigación y la intervención sobre la vida, el medio ambiente y el planeta Tierra. Sin embargo, cabe destacar, que ya en 1978, el Kennedy Institute de la Universidad jesuita de Georgetown en Estados Unidos, había publicado la primera Enciclopedia de Bioética en cuatro volúmenes, dirigida por Warren Reich, un teólogo católico, donde se define a la Bioética como el «estudio sistemático de la conducta humana en el área de las ciencias de la vida y la salud, examinado a la luz de los valores y principios morales».
La bioética abarca las cuestiones éticas acerca de la vida que surgen en las relaciones entre biología, nutrición, medicina, química, política (no debe confundirse con la «biopolítica»), derecho, filosofía, sociología, antropología, teología, etc. Existe un desacuerdo acerca del dominio apropiado para la aplicación de la ética en temas biológicos. Algunos bioéticos tienden a reducir el ámbito de la ética a lo relacionado con los tratamientos médicos o con la innovación tecnológica. Otros, sin embargo, opinan que la ética debe incluir lo relativo a todas las acciones que puedan ayudar o dañar organismos capaces de sentir miedo y dolor. En una visión más amplia, no sólo hay que considerar lo que afecta a los seres vivos (con capacidad de sentir dolor o sin tal capacidad), sino también al ambiente en el que se desarrolla la vida, por lo que también se relaciona con la ecología.
El criterio ético fundamental que regula esta disciplina es el respeto al ser humano, a sus derechos inalienables, a su bien verdadero e integral: la dignidad de la persona.
 articulo bioetica
articulo bioeticaANATOMÍA DE BOCA
La boca es la primera parte del tubo digestivo aunque también se emplea para respirar. Está tapizada por una membrana mucosa, la mucosa oral, con epitelio plano estratificado no queratinizado y limitada por las mejillas y los labios. El espacio en forma de herradura situado entre los dientes y los labios, se llama vestíbulo y el espacio situado por detrás de los dientes es la cavidad oral propiamente dicha. El techo de la cavidad oral está formado por el paladar que consiste en dos partes: una ósea llamada paladar duro, formada por parte de los huesos maxilar superior y palatinos y otra, formada por músculos pares recubiertos de mucosa, llamada el paladar blando o velo del paladar, que se inserta por delante en el paladar duro y, por detrás es libre y presenta una proyección cónica en la línea media, la úvula. A cada lado del paladar blando hay dos músculos recubiertos de repliegues verticales de mucosa que constituyen los dos pilares anteriores y los dos pilares posteriores del paladar y forman el istmo de las fauces o puerta de comunicación de la cavidad oral con la parte oral de la faringe u orofaringe. Entre los pilares, en cada lado, se encuentra una colección de tejido linfoide que constituye las amígdalas palatinas (que cuando se infectan son llamadas popularmente anginas) cuya parte visible no es una guía exacta de su tamaño real porque una gran porción de ellas puede estar oculta por detrás de la lengua. Por su parte anterior la cavidad oral se comunica con el exterior por la abertura de la boca.
ANATOMÍA DE LENGUA
La lengua ocupa la parte media del piso de la boca. Su cara superior está dividida en dos partes (una anterior o bucal y otra posterior o faríngea) por un surco en forma de V abierta hacia adelante llamado surco terminal o V lingual. Está constituida principalmente por músculos intrínsecos (sólo se encuentran en la lengua) y extrínsecos (se originan fuera de la lengua). Entre los músculos intrínsecos está el músculo vertical lingual que desciende la lengua; el músculo transverso lingual, el cual la hace cilíndrica, y el músculo lingual superior que se encuentra en el vértice de la misma. También se encuentra el músculo lingual inferior que llega hasta el ápex del frenillo lingual.
Entre los músculos extrínsecos se encuentra el músculo geniogloso, el cual permite sacar la lengua; el músculo hipogloso, que hace que se retraiga, y el músculo estilogloso, el cual participa en el proceso de la deglución.
La parte faríngea de la cara dorsal de la lengua presenta pequeñas prominencias dispuestas de manera oblicua, se deben a la presencia en la capa superficial de la mucosa de folículos cuyo conjunto constituye las papilas de la V lingual y el foramen caecum, que es el remanente del conducto tirogloso. En el tercio posterior se encuentran las amígdalas linguales.
La inervación motora de la lengua procede del nervio hipogloso (XII) y del glosofaríngeo (IX). La sensación del gusto de los dos tercios anteriores es conducida por la cuerda del tímpano, rama del nervio facial (VII), y la del tercio posterior, por los nervios glosofaríngeo y vago (X). La sensibilidad lingual está dada por la rama lingual de la división mandibular del trigémino (V) y los nervios glosofaríngeo y laríngeo interno.
DIENTES
Los dientes son órganos digestivos accesorios implantados en los alvéolos dentarios situados en los bordes alveolares de la mandíbula y del maxilar superior. En la especie humana aparece primero un grupo de dientes, los dientes de leche o primarios que son temporales. Constan de 2 incisivos, 1 canino y 2 molares (5 piezas) en cada cuadrante. Hay, pues, 20 dientes de leche. Comienzan a aparecer hacia el 6º mes de vida y se completan al final del 2º año.
Alrededor de los 5 años los dientes permanentes sustituyen a los primarios y no se completan hasta después de los 20 años. La dentadura definitiva consta de 8 piezas, en cada cuadrante: 2 incisivos, 1 canino, 2 premolares y 3 molares. Es decir, 32 dientes en total.
Los dientes tienen las siguientes funciones:
Las 2 primeras funciones las realizan los incisivos y caninos porque tienen bordes cortantes. Los premolares y molares que tienen amplias superficies planas, mastican el alimento. Los músculos masticadores, trabajando juntos, pueden cerrar los incisivos con una fuerza de 25 Kg y los molares con una fuerza de 90 Kg.
GLÁNDULAS SALIVARES
La salivación es la secreción de saliva por las glándulas salivares, que en el ser humano es de alrededor de 1 litro por día. Las glándulas salivares están situadas por fuera de las paredes del tubo digestivo. Las más importantes son: las parótidas, las submaxilares y las sublinguales. Son estructuras pares o sea que hay 6 glándulas salivares mayores, aunque existen otras pequeñas.
Las glándulas parótidas están formadas exclusivamente por células serosas que producen una secreción acuosa desprovista de moco. Contribuyen al 25% de la secreción total de saliva en reposo. Cada parótida está situada entre la rama de la mandíbula por delante y la apófisis mastoides por detrás y tiene un conducto que desemboca en la superficie de la mucosa de la mejilla por encima del 2º molar superior. Está atravesada por la arteria carótida externa y el nervio facial.
Las glándulas sublinguales y las glándulas submaxilares están formadas por células mucosas y serosas y situadas por debajo de la mucosa del suelo de la boca, en donde desembocan por varios conductos. Las glándulas submandibulares contribuyen a un 70% de la secreción de saliva en reposo y las sublinguales al restante 5%.
La secreción serosa contiene la amilasa salival o ptialina, un enzima utilizado para digerir el almidón y la secreción mucosa contiene mucoproteínas que dan a la saliva una consistencia pegajosa (moco) y sirve para lubrificar. La saliva basal contiene, además, iones de sodio, cloro y bicarbonato en concentraciones parecidas a las del plasma. La concentración de potasio es superior a la del plasma, de modo que cualquier estado que provoque eliminación excesiva de saliva al exterior dará lugar a una pérdida grave de estos iones.
ESÓFAGO
El esófago es el tubo que conduce el alimento desde la faringe al estómago. Se origina como una continuación de la faringe (a nivel de la VI vértebra cervical) y desciende a través del cuello y el tórax para atravesar después el diafragma (por el hiato esofágico) y alcanzar el estómago. Hasta llegar a la bifurcación de la tráquea, está situado entre la tráquea por delante y la columna vertebral, por detrás.
Después, el pericardio separa el esófago de la aurícula izquierda. Penetra en el estómago formando un ángulo agudo (a nivel de la X vértebra dorsal) y su longitud total es de unos 25 cm. El epitelio de su mucosa es plano estratificado no queratinizado y en las capas musculares de su pared, se encuentra músculo estriado esquelético en su 1/3 superior que gradualmente es sustituido por músculo liso en su 1/3 medio, en donde se encuentran juntas fibras musculares estriadas y lisas, y en su 1/3 inferior ya es músculo liso que se continúa con las capas de músculo liso del estómago.
En la parte superior del esófago existe el esfínter faringoesofágico, entre la faringe y el esófago, que permanece cerrado entre deglución y deglución y por tanto impide que el aire entre en el esófago durante la inspiración y en su extremo inferior, el esfínter gastroesofágico, entre el esófago y el estómago. La función principal de este esfínter es impedir el reflujo del contenido gástrico hacia el esófago, ya que dicho contenido es muy ácido y rico en enzimas proteolíticos y puede dañar la mucosa esofágica que no es capaz de resistir la agresión y se ulcera (esofagitis por reflujo). El diafragma ayuda en la función de este esfínter y también el hecho de que el esófago forme un ángulo agudo al desembocar en el estómago lo que hace más difícil el reflujo.
Irrigación esofágica
El esófago cuenta con tres zonas principales de riego arterial:
Drenaje venoso
Los capilares del esófago fluyen hacia el plexo venoso submucoso y éste drena a un plexo periesofágico. En la región cervical se forman venas esofágicas que drenan en la tiroidea inferior, en la torácica, en las bronquiales, ácigos y hemiácigos y en la porción abdominal en la gástrica izquierda
Drenaje linfático
El esófago cuenta con un drenaje amplio que consiste en vasos linfáticos sumamente interconectados en la mucosa y submucosa esofágicas. De ahí parten hacia la superficie esofágica y se dirigen a los siguientes relevos ganglionares:
La dirección del flujo linfático del esófago es superior a partir del nivel en el que se encuentra la carina traqueal, y la porción esofágica inferior a ella se dirige de forma caudal. Este flujo cefálico se dirige hacia el conducto torácico y el inferior hacia ganglios celiacos.
Inervación
El sistema encargado de la inervación es el nervioso autónomo, y éste lo conforman el sistema nervioso simpático y el sistema nervioso parasimpático. Las fibras simpáticas son eferentes y se encargan de la vasoconstricción, movimientos del tubo digestivo y contracción de los esfínteres. Las parasimpáticas son aferentes y se asocian con la actividad glandular y peristalsis del tubo digestivo.
Además, en el caso del esófago, la inervación se puede dividir en:
ANATOMÍA DE ESTÓMAGO
El estómago es una dilatación del tubo digestivo situada entre el esófago y el duodeno, con una capacidad aproximada de 1-1.5 litros. Difiere del resto del tubo digestivo en que su pared tiene una tercera capa de fibras musculares lisas orientadas de modo oblicuo y situadas en la parte interna de la capa circular. La mayor parte del estómago se encuentra situado en el epigastrio aunque ocupa también parte del hipocondrio izquierdo. Se relaciona por delante con el lóbulo izquierdo hepático y el reborde costal izquierdo, por detrás con el riñón izquierdo, por encima con el diafragma y por debajo con el colon transverso y su mesocolon.
Si consideramos que el estómago tiene forma de J, se puede distinguir una porción vertical y otra horizontal. El pliegue que está entre las dos porciones se llama incisura angular. Un plano que pase por la incisura angular y otro que pase por la unión esófago-gástrica delimitan varias partes:
Irrigación arterial
El estómago tiene un riego anastomótico muy abundante. El aporte sanguíneo proviene del tronco celiaco a través de la gástrica izquierda (coronaria estomática). El aporte sanguíneo de la porción más superior, incluyendo el esófago inferior, proviene de la arteria diafragmática inferior izquierda.
La arteria gástrica izquierda o coronaria estomática tiene 4 a 5 mm. Se origina en el tronco celiaco y discurre por el epiplón menor hasta el cardias, para girar y continuar a lo largo de la curvatura menor del estómago y anastomosarse con la arteria gástrica derecha. Esta última nace de la arteria hepática y se dirige a la izquierda por la curvatura menor, para anastomosarse con la arteria gástrica derecha.
La arteria gastroepiploica derecha se origina como una de las ramas terminales de la arteria gastroduodenal, que es rama de la arteria hepática común. Se dirige a lo largo de la curvatura menor y se anastomosa con la arteria gastroepiploica izquierda.
La arteria gastroepiploica izquierda nace de la arteria esplénica y sigue por la curvatura mayor para anastomosarse con la arteria gastroepiploica derecha. Las arterias gástricas cortas se originan en la porción distal de la arteria esplénica o en las ramas esplénicas, y llegan hasta el fondo del estómago.
Drenaje venoso
Las venas que se originan de la red submucosa e intramuscular siguen el trayecto inverso a las arterias, existe una vena por arteria. La vena gástrica izquierda sigue en sentido inverso el trayecto de la arteria homóloga, describiendo un arco que participa en el pliegue gastropancreático. Llega a nivel del tronco celiaco, sigue a la arteria hepática común y termina en la vena porta. La vena gástrica derecha termina en la vena porta.
La vena gastroepiploica derecha sigue en sentido inverso a la arteria homónima recibiendo a las venas gástricas, epiploicas y subpilóricas y drena en la vena mesentérica superior. La vena gastroepiploica izquierda y las venas gástricas cortas desembocan en la vena esplénica.
En el fondo hay dos grupos de venas, uno derecho y otro izquierdo. El derecho corresponde a la región esofagofúndica y termina en la vena gástrica izquierda. El izquierdo corresponde a las venas gástricas cortas y la gástrica posterior que desembocan en la vena esplénica. Las anastomosis portocavas son numerosas, se ubican en la región esofagogástrica, donde las venas gástricas tributarias de la vena porta se anastomosan con las venas esofágicas, tributarias de la vena cava inferior o de la vena cava superior.
Drenaje linfático
Los vasos linfáticos gástricos acompañan a las arterias a lo largo de las curvaturas mayor y menor del estómago. Drenan la linfa de las caras anterior y posterior y la llevan a las curvaturas, donde se encuentran los ganglios linfáticos gástricos y gastroepiploicos.
Los vasos eferentes de estos ganglios acompañan a las grandes arterias hasta los ganglios linfáticos celiacos. La linfa de los dos tercios superiores del estómago drena por los vasos gástricos derecho e izquierdo hasta los ganglios gástricos; la linfa del fondo y de la parte superior del cuerpo del estómago drena a lo largo de las arterias gástricas cortas y vasos gastroepiploicos izquierdos en los ganglios pancreatoesplénicos.
La linfa de los dos tercios derechos del tercio inferior del estómago drena, a lo largo de los vasos gastroepiploicos derechos, en los ganglios pilóricos. La linfa del tercio izquierdo de la curvatura mayor drena, a lo largo de los vasos gástricos cortos y esplénicos, en los ganglios pancreaticoduodenales.
Inervación
La inervación parasimpática del estómago proviene de los troncos vagales anterior y posterior; sus ramas penetran en el estómago por el hiato esofágico. El tronco vagal anterior deriva del nervio vago izquierdo, penetra al abdomen situado en la cara anterior del esófago, y se dirige a la curvatura menor del estómago en donde emite ramas hepáticas y duodenales que abandonan el estómago con el ligamento hepatoduodenal. El resto del tronco vagal anterior continúa por la curvatura menor, dando ramas gástricas anteriores. El tronco vagal posterior procede del nervio vago derecho.
Entra al abdomen por la cara posterior del esófago y pasa a la curvatura menor del estómago. Da ramas para las caras anterior y posterior del estómago, emite una rama celiaca que se dirige al plexo celiaco y luego continúa por la curvatura menor, dando ramas gástricas posteriores. La inervación simpática del estómago procede de los segmentos T6-T9 de la médula espinal, pasa al plexo celiaco por el nervio esplénico mayor y se distribuye por los plexos que rodean a las arterias gástricas y gastroepiploicas.
Las fibras vagales hacen sinapsis en la pared del estómago con células ganglionares de los plexos mientérico de Auerbach y submucoso de Meissner.
Los nervios que provienen del plexo celiaco son simpáticos y parasimpáticos, dispuestos en forma de plexos arteriales alrededor de las arterias. El píloro está inervado por ramas supra y subpilóricas, provenientes del plexo nervioso de la arteria hepática y de sus ramas (arteria gástrica derecha y gastroepiploica derecha). En la pared gástrica los nervios atraviesan los diferentes planos en compañía de los vasos.
Otras ramas derivan del plexo frénico izquierdo, y llegan al extremo cardial del estómago. También, de forma inconstante, hay ramas que provienen del plexo esplácnico torácico y lumbar
ANATOMIA DE INTESTINO DELGADO
El intestino delgado es un tubo estrecho que se extiende desde el estómago hasta el colon. Consta de 3 partes, duodeno, yeyuno e íleon. El duodeno tiene unos 25 cm de longitud y se extiende desde el píloro hasta el ángulo duodeno-yeyunal, rodeando la cabeza del páncreas. Con fines descriptivos se divide en 3 porciones: primera, segunda y tercera. Igual que sucede con el páncreas, el duodeno está cubierto por peritoneo solamente por su cara anterior, por ello se le considera órgano retroperitoneal. Se relaciona con el estómago, el hígado y el páncreas con los que forma una unidad funcional y recibe el quimo del estómago, las secreciones del páncreas y la bilis del hígado. El colédoco y el conducto pancreático principal desembocan juntos en la segunda porción del duodeno, en la ampolla de Vater o papila duodenal, en donde existe un esfínter, el esfínter de Oddi que está relacionado, sobre todo, con el control del flujo del jugo pancreático al duodeno ya que el flujo de bilis hacia el duodeno está controlado por el esfínter del colédoco situado en el extremo distal de este conducto biliar.
El yeyuno y el íleon tienen en conjunto más de 4.5 m de longitud y debido a que sus características morfológicas y funcionales son parecidas se les puede considerar una unidad: el yeyun-íleon, que forma las llamadas asas del intestino delgado, situadas por debajo del colon transverso y recubiertas por el mesenterio, constituido por pliegues de peritoneo, que las sujeta a la pared abdominal posterior. La desembocadura del íleon en el colon, se produce en el ciego, en el orificio íleocecal a través del cual pasa el contenido del intestino delgado al intestino grueso, y que está rodeado por la válvula íleo-cecal cuya función principal es evitar el reflujo de materias fecales desde el colon al intestino delgado. En los últimos centímetros de íleon, que preceden a la válvula, la pared intestinal posee una pared muscular engrosada, el esfínter íleocecal que, en condiciones normales, se encuentra medianamente contraído y no permite que el contenido del íleon se vacíe en el ciego de un modo brusco y continuado.
Irrigación arterial del duodeno
El duodeno tiene una irrigación sanguínea importante; está irrigado por la arteria celiaca y mesentérica superior. La irrigación principal para el duodeno proviene de las arterias pancreaticoduodenales superior e inferior, ramas de las arterias gastroduodenal y mesentérica superior, respectivamente. La mitad proximal del duodeno está irrigada por la arteria pancreaticoduodenal superior y la distal por la arteria pancreaticoduodenal inferior. Estos vasos se anastomosan formando las arcadas arteriales anterior y posterior.
Drenaje venoso del duodeno
Las venas drenan al sistema venoso portal. La mayoría de las venas duodenales desembocan en la vena mesentérica superior, aunque algunas desembocan directamente en la vena porta. Existen muchas venas pequeñas en la cara anterior y posterior del duodeno, que a veces drenan a las venas pancreaticoduodenales superiores.
Drenaje linfático del duodeno
Los vasos linfáticos de la cara anterior y posterior del duodeno se anastomosan libremente entre sí dentro de la pared duodenal. Los vasos anteriores acompañan a las arterias y drenan cranealmente a los ganglios linfáticos pancreaticoduodenales situados a lo largo de la arteria esplénica y a los ganglios linfáticos pilóricos que se encuentran a lo largo de la arterial gastroduodenal. Los vasos linfáticos posteriores pasan por detrás de la cabeza del páncreas y drenan caudalmente en los ganglios linfáticos mesentéricos superiores, situados alrededor del origen de la arteria mesentérica superior.
Inervación del duodeno
El duodeno está inervado por el nervio vago y los nervios simpáticos, a través de plexos que se localizan en las arterias pancreaticoduodenales.
Irrigación arterial del yeyuno e íleon
Las arterias del yeyuno e íleon proceden de la arteria mesentérica superior, la cual sigue un trayecto oblicuo desde la raíz del mesenterio hasta la fosa iliaca derecha, que emite muchas ramas para el intestino. Estas ramas se unen para formar asas o arcos denominados arcadas arteriales, de los que emergen vasos rectos. Los vasos rectos se dirigen desde las arcadas hasta el borde mesentérico del intestino para llegar al lado contrario. El grado de irrigación del yeyuno es mayor que el del íleon, pero las arcadas arteriales son más cortas y complejas en el íleon.
Drenaje venoso del yeyuno e íleon
La vena mesentérica superior drena el yeyuno e íleon y se une a la vena esplénica para dar lugar a la vena porta.
Drenaje linfático del yeyuno e íleon
Los linfáticos de las vellosidades intestinales, denominados vasos quilíferos, drenan el líquido a un plexo de vasos linfáticos situado en las paredes del yeyuno e íleon. Los vasos linfáticos se dirigen después entre las dos túnicas del mesenterio hacia los ganglios linfáticos mesentéricos superiores. Los vasos linfáticos del íleon terminal acompañan a la rama ileal de la arteria ileocólica y desembocan en los ganglios linfáticos ileocólicos.
Inervación del yeyuno e íleon
El intestino delgado tiene inervación simpática y parasimpática. Las fibras parasimpáticas provienen del vago y atraviesan los ganglios celiacos. Las simpáticas provienen de los tres grupos de nervios esplácnicos cuyas células ganglionares se encuentran en un plexo alrededor de la base de la arteria mesentérica superior. El dolor intestinal es mediado a través de fi bras aferentes viscerales generales del sistema simpático.
INTESTINO GRUESO
El intestino grueso se extiende desde la válvula íleo-cecal hasta el ano y tiene unos
1.5 m de longitud.
Consta de: ciego, apéndice, colon ascendente, colon transverso, colon descendente, colon sigmoide, recto y conducto anal.
FISIOLOGÍA
REFLEJO DE MASTICACIÓN. FUNCIONES
La masticación es la primera fase de la digestión y se realiza en la boca, utilizando dos tipos de dientes, los premolares y los molares. Consiste en la conversión de las partículas grandes de alimento en otras más pequeñas, de fácil deglución. Una gran parte del proceso de masticación está causado por el reflejo de masticación, que consiste en lo siguiente:
Las funciones de la masticación son:
SALIVACIÓN. REGULACIÓN. FUNCIONES
En la boca, el alimento se fragmenta en trozos más pequeños por la masticación y se mezcla con saliva. La presencia de alimento en la boca y los estímulos sensoriales de gusto y olfato tienen una función importante en la estimulación de la secreción de la saliva. En reposo se secretan 0.5 ml de saliva por minuto que se pueden incrementar hasta 7 ml por minuto debido a determinados alimentos, olores o al propio proceso de masticación. La secreción de saliva está regulada por reflejos mediados por el sistema nervioso simpático y el sistema nervioso parasimpático. La estimulación parasimpática origina la secreción de una saliva rica en amilasa y mucina, con aumento de la secreción de bicarbonato. La respuesta de la secreción de saliva al estímulo simpático es variable aunque el resultado neto es una disminución de la secreción de saliva. La boca seca es una característica importante de la respuesta simpática al miedo o al estrés. Las glándulas es producen cada día de promedio, unos 1500 ml de saliva cuyas funciones son:
DEGLUCIÓN
Una vez que el alimento ha sido masticado y mezclado con la saliva se forma un bolo alimenticio que puede ser tragado. El acto de tragar es la deglución. En la deglución, el bolo pasa por tres espacios: la boca, la faringe y el esófago. Por ello, se distinguen tres etapas en la deglución:
El esfínter gastroesofágico actúa como una válvula ya que permanece cerrado cuando no se está deglutiendo ningún alimento para evitar la regurgitación de jugo gástrico pero, justo antes de que la onda peristática alcance el final del esófago, se relaja para permitir la entrada del bolo al interior del estómago. Además la submucosa del esófago contiene glándulas que secretan moco en respuesta a la presión provocada por el bolo y ayuda a lubrificar el esófago y facilitar el transporte del alimento.
La velocidad de paso del bolo alimenticio por el esófago depende, sobre todo, de la consistencia del bolo y de la postura del cuerpo. En posición erecta en que la gravedad ayuda, el agua alcanza el estómago en 1 segundo, un contenido en forma de papilla en 5 segundos y las partículas sólidas en 9-10 segundos o más.
ESTÓMAGO. FUNCIONES
Las funciones del estómago son:
ESTÓMAGO. VACIAMIENTO
Cuando entra el bolo alimenticio en el estómago, se va disponiendo en el cuerpo del estómago en forma concéntrica, desplazando hacia la periferia el alimento que ya estaba en la cavidad. Por esta razón continúa durante un tiempo la actividad de la amilasa salivar antes de que el jugo gástrico entre en contacto con el bolo alimenticio y detenga su acción. Las secreciones gástricas actúan en la parte del alimento almacenado que se encuentra situado en contacto con la mucosa del estómago.
Cuando el estómago contiene alimento, se producen ondas constrictoras débiles llamadas ondas de mezclado que se mueven a lo largo de su pared aproximadamente 1 vez cada 20 segundos Estas ondas dan lugar a que las secreciones gástricas se mezclen bien con el alimento almacenado y además tienen un efecto propulsor que va moviendo el contenido gástrico hacia el antro pilórico. A medida que las ondas constrictoras del cuerpo del estómago progresan hasta el antro se hacen más intensas y algunas son muy potentes y se extienden por el antro permitiendo la salida del quimo por el esfínter pilórico. Como la abertura del esfínter es muy pequeña, solo son vaciados hacia el duodeno unos pocos ml de quimo con cada onda y a continuación el esfínter se cierra de inmediato. El contenido del antro que no puede atravesar el esfínter pilórico es empujado de nuevo hacia el cuerpo del estómago, lo que constituye un mecanismo de mezcla muy importante. Al irse vaciando cada vez más el estómago, las contracciones peristálticas llegan más arriba en el cuerpo y van mezclando y fragmentando las porciones más recientes del alimento almacenado.
El líquido salino isotónico y el agua son las sustancias vaciadas más rápidamente por el estómago, sin retardo y más rápido cuanto más volumen de líquido. Los líquidos ácidos dejan el estómago más lentamente. En cuanto a los sólidos, el vaciamiento gástrico varía con el tamaño de las partículas (los componentes sólidos pasan al píloro solo si se han triturado a un tamaño de 2-3 mm, abandonando el estómago en un 90% con un tamaño de 0.25 mm) y el tipo de alimento. Los primeros en abandonar el estómago son los carbohidratos, después las proteínas y, por último, las grasas que pueden tardar hasta 4 horas.
ESTÓMAGO. VACIAMIENTO. REGULACIÓN
El vaciamiento gástrico depende de la fuerza de las ondas peristálticas del antro pilórico y del grado de contracción del esfínter pilórico que son controlados, a su vez, por señales reguladoras procedentes del estómago y del intestino delgado. Las funciones fundamentales de la unión gastroduodenal son: permitir el vaciamiento cuidadosamente regulado del contenido gástrico a un ritmo compatible con la capacidad del duodeno para procesar el quimo y evitar el reflujo del contenido duodenal hacia el estómago.
Señales reguladoras que provienen del estómago. Son de 2 tipos:
Señales hormonales: se deben a unas hormonas secretadas por células de la mucosa del intestino delgado en respuesta a ciertos componentes del quimo. Las más importantes son la secretina por la mucosa del duodeno y la colecistoquinina y el péptido inhibidor gástrico por la mucosa del duodeno y del yeyuno proximal. Estas hormonas, una vez secretadas, pasan a la sangre y actúan sobre el estómago para inhibir la fuerza de contracción del antro y aumentar el tono del esfínter pilórico, con lo que enlentecen el vaciamiento gástrico. Los componentes del quimo que estimulan la secreción de estas hormonas son: los ácidos (la secretina se libera a la sangre cuando el pH del duodeno desciende por debajo de 4.5) y los productos de desintegración de las grasas (la colecistoquinina y el péptido inhibidor gástrico).
ESTÓMAGO. SECRECIÓN
El estómago secreta diariamente de 2-3 litros de jugo gástrico. La mucosa gástrica presenta varios tipos de células y glándulas secretoras:
ESTÓMAGO. SECRECIÓN. REGULACIÓN
La ingesta de alimentos es el estímulo adecuado para la estimulación de la secreción del jugo gástrico que comienza ya antes de la comida y sigue después de terminarla. En la secreción gástrica se distinguen 3 fases, la fase cefálica, gástrica e intestinal que se solapan en el tiempo.
Señales hormonales: la gastrina, a su vez, actúa sobre las glándulas gástricas dando lugar a más producción de jugo gástrico muy ácido (es decir, con un contenido muy elevado de ClH). La gastrina no solo se secreta por estímulo parasimpático sino también por la presencia de ciertos alimentos en el antro, como los péptidos y aminoácidos libres, que tienen un efecto estimulante directo sobre su secreción. Un medio muy ácido con un pH por debajo de 3 en el antro pilórico inhibe la liberación de gastrina. En condiciones normales, al llegar alimento al estómago, se produce un aumento de pH con lo que se hace menos ácido y la gastrina se libera y, como consecuencia, se va secretando jugo gástrico hasta que llega un momento en que el pH se hace más ácido y entonces se inhiben las células productoras de gastrina y deja de secretarse. La inhibición de la secreción de gastrina está mediada por un aumento de la secreción de somatostatina por las células D de la mucosa del antro pilórico. De este modo se autolimita la secreción gástrica.
INTESTINO DELGADO. MOTILIDAD
El quimo atraviesa todo el intestino delgado en unas 3-5 horas, aunque puede ser en más tiempo. Los pliegues circulares de la mucosa intestinal, debido a su forma, fuerzan al quimo a seguir un trayecto en espiral a medida que va avanzando. Este movimiento en espiral enlentece el movimiento del quimo y facilita el mezclado con los líquidos intestinales, optimizando las condiciones para la digestión y la absorción. Los alimentos de los que vive el organismo, con la excepción de pequeñas cantidades de sustancias como vitaminas y minerales, pueden ser clasificados como carbohidratos, proteínas y grasas. Estos alimentos tal como son ingeridos, no pueden atravesar las células intestinales y, por tanto, deben desdoblarse en moléculas más sencillas, mediante la digestión, para poder ser absorbidas después a través de la pared del intestino delgado. La absorción intestinal consiste en el paso de los productos resultantes de la digestión a través de las células epiteliales de la mucosa del intestino delgado para llegar a la sangre de la vena porta o a la linfa. Cada día se absorben por el intestino delgado varios cientos de gramos de carbohidratos, 100 o más gramos de grasas, 50-100 gramos de aminoácidos, 50-100 gramos de iones y 7-8 litros de agua.
En el intestino delgado se producen 2 tipos de movimientos que tienen los siguientes objetivos:
INTESTINO DELGADO. VACIAMIENTO. REGULACIÓN
El vaciamiento del intestino delgado es regulado a partir de señales reguladoras procedentes del estómago y señales reguladoras procedentes del ciego.
INTESTINO DELGADO. SECRECIÓN. REGULACIÓN
El jugo intestinal es la mezcla de las secreciones de las siguientes células y glándulas:
DIGESTIÓN Y ABSORCIÓN DE HIDRATOS DE CARBONO
El consumo diario de hidratos de carbono en las dietas occidentales es de unos 250-800 g. Casi todos los carbohidratos de la dieta son grandes polisacáridos o disacáridos que son combinaciones de monosacáridos. Hay 3 fuentes principales de carbohidratos en la dieta normal: la sucrosa, disacárido conocido como azúcar de caña; la lactosa, disacárido de la leche y los almidones, grandes polisacáridos presentes en casi todos los alimentos. El almidón vegetal o amilopectina es la principal fuente de hidratos de carbono en la mayoría de las dietas humanas. La cantidad ingerida del almidón animal o glucógeno, varía mucho según las culturas. Junto a ellos se consumen pequeñas cantidades de monosacáridos como la glucosa y la fructosa.
La digestión de los polisacáridos comienza en la boca por la acción de la amilasa salivar que continúa actuando durante el paso por el esófago y en el estómago hasta que se inactiva por el descenso de pH, al entrar en contacto con el jugo gástrico. La amilasa de la saliva puede disociar el almidón hasta el 50% si se mastica durante un tiempo suficiente y sigue trabajando en el bolo alimenticio ya que la estratificación en capas concéntricas en el estómago impide su inactivación. En el duodeno, la digestión del almidón se realiza muy rápido por la amilasa pancreática.
Pero como los carbohidratos solo pueden absorberse en forma de monosacáridos, los productos resultantes de la digestión por las amilasas, que son oligosacáridos, tienen que seguir desintegrándose. Esto lo realizan las disacaridasas de la membrana de las microvellosidades de las células epiteliales columnares del duodeno y yeyuno.
Todos los hidratos de carbono son convertidos al final en monosacáridos: fructosa, galactosa y glucosa. La glucosa y la galactosa son absorbidas, entrando en las células epiteliales por la membrana del borde en cepillo, en contra de gradiente utilizando un mecanismo de cotransporte dependiente de sodio, y saliendo de las células por sus membranas plasmáticas basal y lateral por difusión facilitada, pasando a la sangre de la vena porta para llegar al hígado. La fructosa no puede ser transportada en contra de gradiente, de modo que se absorbe desde la luz intestinal al interior de las células epitaliales mediante difusión facilitada independiente del sodio. El duodeno y el yeyuno proximal poseen la mayor capacidad para absorber azúcares, que resulta menor en el yeyuno distal y el íleon.
DIGESTIÓN Y ABSORCIÓN DE GRASAS
La ingesta diaria de grasas es de 60-100 g. Las grasas más comunes de la dieta son las grasas neutras o triglicéridos (la inmensa mayoría formados por ácidos grasos de cadenas largas). También hay pequeñas cantidades de colesterol, fosfolípidos y vitaminas liposolubles. En el estómago, los lípidos forman grandes gotas de grasa. Lo primero que pasa cuando las grasas llegan al duodeno es que las sales biliares recubren las gotas de grasa y éstas se rompen, dividiéndose en gotitas de grasa más pequeñas que aumentan miles de veces la superficie de actuación de los enzimas lipolíticos del páncreas. Este proceso se llama emulsión de las grasas (una emulsión es una suspensión acuosa de pequeñas gotas de grasa) y permiten el acceso a los triglicéridos de la lipasa pancreática que los rompe en monoglicéridos y ácidos grasos. De este modo los movimientos gastrointestinales pueden romper las gotas de grasa en partículas más y más finas. Si no hubiese bilis, todos los lípidos se unirían formando un gran globo de grasa, exponiendo la menor superficie posible al agua. Como los enzimas pancreáticos son hidrosolubles, solo actuarían en la superficie del globo de grasa expuesta al agua y la digestión de las grasas sería mínima.
A su vez, los productos de la digestión de los lípidos, junto con colesterol, fosfolípidos y vitaminas liposolubles, forman pequeños agregados moleculares con las sales biliares que se llaman micelas, con la cara hidrofóbica orientada hacia el interior lipídico de la micela y la cara hidrofílica hacia el exterior. El tamaño de las micelas es lo bastante pequeño como para difundir entre las microvellosidades y permitir la absorción de los lípidos por la membrana plasmática del borde en cepillo de las células epiteliales intestinales. Una vez dentro del citoplasma celular, los monoglicéridos y los ácidos grasos se resintetizan de nuevo en triglicéridos en el retículo endoplasmático liso que, asociados con colesterol y vitaminas liposolubles y rodeados de fosfolípidos y una lipoproteína forman los quilomicrones que son expulsados de la células epiteliales intestinales por exocitosis y pasan a los espacios intercelulares laterales, entrando en los capilares linfáticos ya que los quilomicrones son demasiado grandes para atravesar la membrana de los capilares sanguíneos.
Los quilomicrones abandonan el intestino con la linfa, que los transportará a la circulación sanguínea general.
El duodeno y el yeyuno son los segmentos más activos en la absorción de las grasas, de modo que la mayor parte del total ingerido ya se ha absorbido cuando el quimo llega al yeyuno medio. Las grasas presentes en las heces normales no proceden de la alimentación, que se absorben por completo, sino de las bacterias del colon y de células intestinales exfoliadas.
Una vez que sueltan a los productos de digestión de las grasas, las sales biliares vuelven al quimo para ser usadas una y otra vez para este proceso de transporte de lípidos en las micelas, hasta que se reabsorben en el íleon distal y son recicladas por los hepatocitos cuando llegan al hígado por la circulación enterohepática.
DIGESTIÓN Y ABSORCIÓN DE PROTEÍNAS
Las personas adultas ingieren diariamente 70-90 g de proteínas. La digestión de las proteínas comienza en el estómago por la pepsina que convierte a las proteínas en grandes polipéptidos. Este enzima funciona solamente a pH muy ácido. Solo un 10-20% de proteínas se digiere en el estómago. El resto en el intestino delgado. La pepsina es especialmente importante por su habilidad para digerir el colágeno que no es afectado por los otros enzimas. Ya que el colágeno es un constituyente importante de la carne, es esencial que sea digerido para que el resto de la carne pueda ser atacado por los otros enzimas digestivos.
Luego el resto de las proteínas es digerido en el intestino delgado por la acción de enzimas proteolíticos pancreáticos como la tripsina. Las proteasas pancreáticas son muy activas en el duodeno y convierten rápidamente las proteínas ingeridas en péptidos pequeños. Alrededor del 50% de las proteínas de la dieta se digieren y absorben en el duodeno. El borde en cepillo de las células epiteliales del duodeno y del intestino delgado contiene, a su vez, diversas peptidasas. El resultado final de la acción de las proteasas pancreáticas y estas peptidasas son péptidos pequeños y aminoácidos simples.
Estos pequeños péptidos y los aminoácidos se transportan a través del borde en cepillo de la membrana apical de las células epiteliales intestinales hacia el citoplasma de las mismas. La velocidad de transporte de los dipéptidos o tripéptidos suele ser mayor que la de los aminoácidos aislados. Los aminoácidos son absorbidos en el ribete en cepillo de las células epiteliales intestinales mediante un mecanismo de cotransporte dependiente de sodio, similar al que se utiliza para la absorción de los monosacáridos. Existen 10 transportadores diferentes para los aminoácidos, de los que siete se localizan en la membrana del ribete en cepillo y tres en la membrana basolateral. Por su parte, los péptidos de pequeño tamaño entran en las células epiteliales utilizando un transportador que no está ligado al sodio sino a los iones H+. En el citoplasma celular los pequeños péptidos son convertidos en aminoácidos simples por peptidasas citoplasmáticas. Los aminoácidos entonces salen de las células epiteliales intestinales por un sistema transportador de aminoácidos que existe en la superficie basolateral y por la sangre de la vena porta llegan al hígado que, por consiguiente, solo recibe aminoácidos simples.
Si la comida se ha masticado bien y en una pequeña cantidad cada vez, alrededor del 98% de las proteínas ingeridas se convierte en aminoácidos y es absorbida y solo el 2% es eliminada en las heces. En las personas normales, casi todas las proteínas de la dieta ya están digeridas y absorbidas en el momento de llegar el quimo a la zona intermedia del yeyuno.
INTESTINO GRUESO. SECRECIÓN. FORMACIÓN Y COMPOSICIÓN DE LAS HECES
Aproximadamente unos 500 ml de quimo pasan cada día desde el íleon al ciego. La mucosa del intestino grueso es lisa ya que no tiene vellosidades y el ribete en cepillo de sus células epiteliales columnares no contiene enzimas. Hay gran cantidad de células caliciformes productoras de moco dispersas entre las células columnares. Por tanto, la secreción del intestino grueso consiste en un líquido mucoso, conteniendo grandes cantidades de iones bicarbonato, y su misión consiste en: evitar lesiones a la mucosa, asegurar la cohesión del bolo fecal y proteger la mucosa contra la intensa actividad bacteriana de esta zona. Cuando una zona del intestino grueso está muy irritada, la mucosa secreta además de moco, grandes cantidades de agua y electrolitos. De este modo se diluyen las sustancias irritantes y se acelera el tránsito de las heces hacia el ano, dando lugar a una diarrea.
La absorción de carbohidratos, lípidos y proteínas, así como de otros nutrientes ya se ha completado en el momento en que el quimo pasa el esfínter íleocecal. De modo que el quimo que pasa al intestino grueso contiene restos celulares, fibras y grandes cantidades de agua y electrolitos. La mayor parte del agua y los electrolitos contenidos en este quimo, se absorben en el colon por lo que quedan menos de 100 ml de líquido para ser excretados en las heces. Toda la absorción que tiene lugar en el intestino grueso ocurre en su 1/2 proximal por lo que a esta parte se le llama colon de absorción. La 1/2 distal tiene como misión almacenar las materias fecales por lo que se llama colon de almacenamiento.
El colon proximal tiene muchas bacterias (hay más de 400 tipos de bacterias en el colon, algunas anaerobias y otras aerobias) que constituyen la flora bacteriana intestinal. La flora intestinal realiza varias funciones:
INTESTINO GRUESO. REFLEJO DE LA DEFECACIÓN
El colon presenta movimientos de mezclado y movimientos propulsores lentos. Las ondas peristálticas se producen varias veces al día y sirven para mover el contenido del intestino grueso en largas distancias. El recto permanece habitualmente vacío y el conducto anal esta cerrado por los esfínteres anales, de modo que la coordinación del recto y el conducto anal es importante para la defecación. Después de la entrada de los alimentos en el estómago, la motilidad del colon aumenta debido al reflejo gastrocólico. Cuando las heces llegan al recto se desencadena el reflejo de la defecación que comienza con la distensión del recto por las heces. Como consecuencia, se inician ondas peristálticas en el colon descendente, el colon sigmoide y el recto que fuerzan las heces hacia el ano. Al aproximarse la onda peristáltica al ano se inhibe el esfínter anal interno, que es involuntario. Si también se relaja el esfínter anal externo se produce la defecación.
Pero este esfínter, al contrario del anterior, puede controlarse voluntariamente y si se mantiene contraído no se produce la defecación. De modo que si se mantiene contraído voluntariamente el esfínter externo, el reflejo de defecación se disipa al cabo de unos minutos y se mantiene inhibido durante horas o hasta que entran más heces en el recto. Las personas que inhiben con demasiada frecuencia el reflejo natural de la defecación, acaban sufriendo estreñimiento. Normalmente se eliminan unos 100-150 gramos de heces cada día.
Histología de esófago
El esófago se compone de cuatro partes o capas:
Histología de estomago
Cuando se abre la víscera mediante un corte a través del plano que delimita sus dos curvaturas, se pueden observar dos segmentos; una porción globulosa hacia la izquierda y una porción estrecha y tubular hacia la derecha. La transición entre estas dos regiones es gradual y su división es puramente arbitraria.
La escotadura cardial se encuentra a la izquierda de la porción abdominal del esófago, su proyección dentro de la cavidad aumenta a medida que el órgano se dilata, por lo que se ha supuesto que actúa como válvula que evita la regurgitación hacia el esófago. La elevación correspondiente a la escotadura pilórica se reconoce señalando el comienzo de la región pilórica, cuyo final viene marcado por el engrosamiento circular del esfínter pilórico.
Los moldes realizados del interior del estómago han demostrado la existencia de un canal (canal gástrico) que se extiende a lo largo de la curvatura menor, desde el cardias hasta la escotadura pilórica.
La estructura de la pared gástrica del estómago, de afuera hacia adentro, está formada por cuatro capas: serosa, muscular, submucosa y mucosa.
La mucosa del esófago distal tiene un grueso epitelio escamoso estratificado, que cambia de forma abrupta a nivel del cardias a un epitelio regular simple de células epiteliales columnares y altas que secretan moco. La mucosa del estómago tiene arrugas, pliegues; en la región de curvatura menor la mucosa forma 4 a 5 pliegues longitudinales paralelos, en el resto las arrugas están dispuestas con menos regularidad, en toda la superficie hay pequeños mamelones de hasta 6 mm de diámetro.
La submucosa está formada por tejido areolar con fibras elásticas y grasa, contiene un extenso plexo de vasos sanguíneos y nervios.
La capa muscular está situada inmediatamente debajo de la serosa; tiene tres estratos, interno oblicuo, medio circular y externo longitudinal
Histología de intestino delgado
La pared del intestino delgado tiene cuatro capas: mucosa, submucosa, muscular y serosa.
Mucosa
La mucosa del intestino delgado se encuentra diseñada para incrementar la superficie de absorción intestinal hasta casi 200 m2. La mucosa se divide en tres capas: epitelio, lámina propia y muscular de la mucosa.
La muscular de la mucosa es una hoja delgada de músculo que separa la mucosa de la submucosa. La lámina propia es una capa continua de tejido conjuntivo entre el epitelio y la muscular de la mucosa; se extiende dentro de las vellosidades y alrededor de las criptas en foso de Lieberkühn. La lámina propia contiene varias células plasmáticas, macrófagos, linfocitos, cebadas, eosinófilos, músculo liso, fibroblastos y tejido conjuntivo no celular. En la lámina propia se producen varios mediadores (citocinas) que modulan diversas funciones celulares; además, las células plasmáticas producen inmunoglobulinas.
Las vellosidades tienen una longitud de 0.5 a 1mm hacia la luz, son más altas en el duodeno distal y en el yeyuno proximal, y se acortan de manera progresiva hacia el íleon terminal. Millones de vellosidades le dan al intestino delgado un aspecto aterciopelado. Cada vellosidad contiene una arteriola, una vénula y un vaso linfático (quilífero). Las vellosidades se continúan con las criptas intestinales o de Lieberkühn, cada una con tipos celulares diferentes.
Las funciones principales del epitelio de las criptas son la renovación celular y la secreción exocrina, endocrina, de agua y electrólitos; la función principal de epitelio de las vellosidades es la digestión y absorción. Las criptas tienen cuatro tipos celulares:
A medida que se producen nuevas células, las células viejas son empujadas hacia arriba y hacia afuera de cada cripta, moviéndose hasta el extremo distal de cada vellosidad, así, la mucosa intestinal está en constante renovación. Se dice que este recambio toma de cinco a siete días en el intestino delgado proximal, mientras que en el íleon tarda tres días. El epitelio que cubre las vellosidades está constituido por células caliciformes, células endocrinas y de absorción (enterocitos).
Las funciones de absorción y digestión son realizadas por los enterocitos, con superficie luminal cubierta con hasta 1 700 microvellosidades por célula, lo que constituye el llamado borde en cepillo, y con lo que se logra multiplicar 30 veces la superficie celular de absorción; además, las enzimas digestivas del intestino delgado están incrustadas en la membrana celular de las microvellosidades.
Submucosa
Es una capa de tejido conjuntivo fi broelástico que contiene vasos sanguíneos y nervios. Es el componente más fuerte de la pared del intestino delgado. Contiene redes complicadas de linfáticos, arteriolas y vénulas, así como un plexo extenso de fibras nerviosas y células ganglionares (plexo de Meissner). En la submucosa del duodeno (pero no en el yeyuno ni íleon) se encuentran las glándulas de Brunner, que se encargan de secretar líquido alcalino, rico en bicarbonato y que contiene moco, el cual protege la mucosa duodenal de la erosión por el ácido y la pepsina del quimo que llega desde el estómago.
Muscular
Esta capa consiste en una porción delgada longitudinal externa y una circular interna más gruesa de músculo liso. Entre las dos capas musculares se encuentran interpuestas las células ganglionares del plexo mientérico de Auerbach y envían fi bras a ambas.
Serosa
Es la capa más externa y consiste en peritoneo visceral que circunda el yeyuno-íleon, pero cubre el duodeno sólo en la parte anterior. Está constituido por una capa de células mesoteliales aplanadas.
Histología del intestino grueso
El hipotiroidismo es un síndrome clínico que se produce por una deficiencia de hormonas tiroideas, que a su vez da lugar a una lentificación generalizada de los procesos metabólicos. El hipotiroidismo en lactantes y niños da por resultado lentificación notoria del crecimiento y desarrollo, con consecuencias permanentes graves, incluso retraso mental, cuando ocurre durante la lactancia. El hipotiroidismo con inicio durante la adultez causa un decremento generalizado del metabolismo, con lentificación de la frecuencia cardiaca, consumo de oxígeno disminuido, y depósito de glucosaminoglucanos en espacios intercelulares, particularmente en la piel y el músculo, lo que en casos extremos produce el síndrome clínico de mixedema. Los síntomas y signos de hipotiroidismo en adultos son reversibles con terapia.
Etiología e incidencia
El hipotiroidismo puede clasificarse como: 1) primario (insuficiencia tiroidea) (con mucho el más frecuente); 2) secundario (debido a deficiencia de TSH hipofisaria), o 3) terciario (debido a deficiencia hipotalámica de TRH), o puede deberse a 4) resistencia periférica a la acción de hormonas tiroideas. El hipotiroidismo también puede clasificarse como con bocio o sin bocio, pero esta clasificación probablemente es insatisfactoria, porque la tiroiditis de Hashimoto (tiroiditis autoinmunitaria) puede producir hipotiroidismo con un bocio o sin éste.
La incidencia de las diversas causas de hipotiroidismo varía dependiendo de factores geográficos y ambientales, como ingestión de yoduro y de bociógenos en la dieta, las características genéticas de la población, y la distribución de la población por edades (pediátrica o adulta). La tiroiditis de Hashimoto es con mucho la causa más común de hipotiroidismo en el mundo desarrollado. En pacientes más jóvenes es más probable que se relacione con bocio; en pacientes de mayor edad, el proceso inmunitario puede destruir por completo la glándula, y el único rastro de la enfermedad son resultados persistentemente positivos en una prueba para anticuerpos contra TPO. De modo similar, la etapa terminal de la enfermedad de Graves puede ser hipotiroidismo, que ocurre de modo espontáneo o después de terapia destructiva con yodo radiactivo o tiroidectomía. Las glándulas tiroides afectadas por enfermedad autoinmunitaria son en particular susceptibles a ingestión excesiva de yoduro (p. ej., ingestión de tabletas de algas, en especial del género Laminaria, preparaciones para la tos que contienen yoduro o el fármaco antiarrítmico amiodarona), o administración intravenosa de medios de contraste radiográficos que contienen yoduro.
Las cantidades grandes de yoduro bloquean la síntesis de hormona tiroidea mediante el efecto de Wolff-Chaikoff, lo que produce hipotiroidismo inducido por yodo, con bocio, en el paciente con una glándula tiroides anormal; la glándula normal escapa al efecto de Wolff-Chaikoff o bloqueo por yoduro, pero por razones que no están claras la autoinmunidad hace a la glándula más sensible a los efectos inhibidores del yodo. Puede ocurrir hipotiroidismo durante la fase tardía de tiroiditis subaguda o de tiroiditis silenciosa; esto por lo general es transitorio, pero puede ser permanente, especialmente en la tiroiditis silenciosa, en la cual se observa hipotiroidismo permanente en alrededor de 25% de enfermos. La deficiencia de yodo no es una causa de hipotiroidismo en Estados Unidos, pero aún se observa en países en desarrollo, y es la causa más común de hipotiroidismo en todo el mundo. Ciertos fármacos pueden bloquear la síntesis de hormona y producir hipotiroidismo con bocio; en la actualidad, las causas farmacológicas más comunes de hipotiroidismo (que no son yoduro) son carbonato de litio, usado para el tratamiento de enfermedad bipolar, y amiodarona. La terapia crónica de hipertiroidismo con los fármacos antitiroideos PTU y metimazol tienen los mismos efectos.
El interferón alfa, usado para tratar hepatitis C y otras enfermedades, puede causar inmunidad alterada que puede dar por resultado hipotiroidismo debido a tiroiditis de Hashimoto. Los errores congénitos de la síntesis de hormona tiroidea, llamados dishormonogénesis tiroidea, se producen por deficiencias genéticas de enzimas necesarias para la biosíntesis de hormona. Estos efectos pueden ser completos, lo que da lugar a un síndrome de hipotiroidismo congénito grave (cretinismo) con bocio, o parciales, lo que suscita bocio con hipotiroidismo más leve. Se han reportado al menos cinco anormalidades biosintéticas separadas: 1) alteración del transporte de yodo; 2) peroxidasa tiroidea deficiente con oxidación alterada de yodo, y fracaso de la incorporación de yodo hacia la tiroglobulina; 3) acoplamiento alterado de tirosinas yodadas a triyodotironina o tetrayodotironina; 4) falta de yodotirosina desyodasa o deficiencia de la misma, de modo que el yodo no se conserva dentro de la glándula, y 5) producción excesiva de yodo proteína inactiva desde el punto de vista metabólico por la glándula tiroides. Esto último puede comprender síntesis alterada o anormal de tiroglobulina.
Las deficiencias hipofisarias e hipotalámicas como causas de hipotiroidismo son bastante raras, y por lo general se relacionan con otros síntomas y signos de insuficiencia hipofisaria.
Patogenia
La deficiencia de hormona tiroidea afecta a casi cualquier tejido del cuerpo, de modo que los síntomas son múltiples. Desde el punto de vista patológico, el dato más característico es la acumulación de glucosaminoglucanos, en su mayor parte de ácido hialurónico en tejidos intersticiales. La acumulación de esta sustancia hidrofílica y el aumento de la permeabilidad capilar a la albúmina explican el edema intersticial sin signo de Godete que es en particular evidente en la piel, el músculo cardiaco y el músculo estriado. La acumulación no se debe a síntesis excesiva de glucosaminoglucanos, sino a metabolismo disminuido de los mismos.
Presentaciones clínicas y datos
 habitual en la parte anterior baja del cuello, lo que da por resultado falta de glándula tiroides o glándula tiroides ectópica que funciona mal. La transferencia placentaria hacia el embrión de anticuerpos bloqueadores contra receptor de TSH desde una madre con tiroiditis de Hashimoto puede dar por resultado agenesia de la glándula tiroides y cretinismo atirótico, pero por lo general sólo causa hipotiroidismo transitorio. Los defectos hereditarios de la biosíntesis de hormona tiroidea también pueden causar hipotiroidismo y bocio neonatales. Otras causas posibles de hipotiroidismo neonatal son exposición a yoduros durante el embarazo, administración de fármacos antitiroideos a la madre, o administración inadvertida de yodo radiactivo para tirotoxicosis o cáncer tiroideo.
habitual en la parte anterior baja del cuello, lo que da por resultado falta de glándula tiroides o glándula tiroides ectópica que funciona mal. La transferencia placentaria hacia el embrión de anticuerpos bloqueadores contra receptor de TSH desde una madre con tiroiditis de Hashimoto puede dar por resultado agenesia de la glándula tiroides y cretinismo atirótico, pero por lo general sólo causa hipotiroidismo transitorio. Los defectos hereditarios de la biosíntesis de hormona tiroidea también pueden causar hipotiroidismo y bocio neonatales. Otras causas posibles de hipotiroidismo neonatal son exposición a yoduros durante el embarazo, administración de fármacos antitiroideos a la madre, o administración inadvertida de yodo radiactivo para tirotoxicosis o cáncer tiroideo.Los signos de hipotiroidismo en recién nacidos son dificultad respiratoria, cianosis, ictericia, alimentación inadecuada, llanto ronco, hernia umbilical, y retraso notorio de la maduración ósea. La epífisis tibial proximal y la epífisis femoral distal están presentes en la mayoría de los lactantes a término con un peso corporal de más de 2 500 g.
La falta de estas epífisis sugiere fuertemente hipotiroidismo. La introducción de pruebas de detección sistemáticas de recién nacidos para TSH o T4 en el mundo desarrollado ha sido un importante logro de salud pública, porque el diagnóstico temprano puede prevenir retraso mental permanente. Una gota de sangre obtenida mediante punción del talón 24 a 48 h después del nacimiento se coloca sobre papel filtro y se envía a un laboratorio central. Una T4 sérica de menos de 6 μg/ dl o una TSH sérica de más de 25 mU/L es sugestiva de hipotiroidismo neonatal. A continuación el diagnóstico puede confirmarse al repetir las pruebas y al obtener evidencia radiográfica de edad ósea retardada. Nótese que los lactantes eutiroideos hijos de madres hipotiroideas que han recibido tratamiento insuficiente con levotiroxina durante el embarazo no son hipotiroideos, pero pueden tener potencial intelectual disminuido en etapas más avanzadas de la niñez, lo que recalca la importancia de mantener a la madre en un estado eutiroideo durante todo el embarazo.
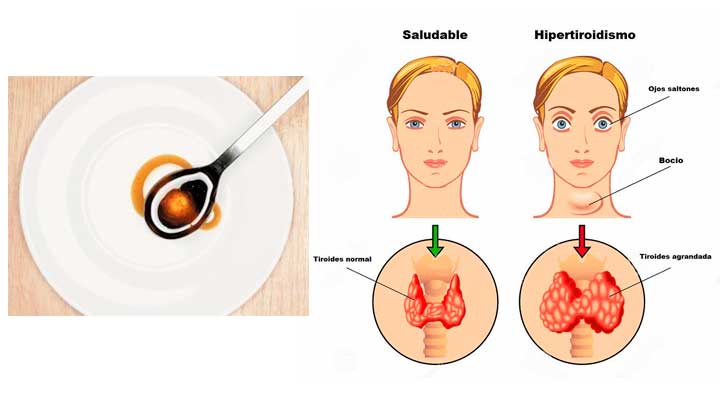
Los datos físicos pueden incluir piel fría, áspera y reseca; cara y manos abotagadas, voz ronca y reflejos lentos. La conversión reducida de caroteno en vitamina A, y la concentración sanguínea aumentada de caroteno, pueden impartir un color amarillento a la piel. Sin embargo, muchos de los síntomas y signos, o todos, están disminuidos o no se observan en pacientes con grados más leves de insuficiencia tiroidea.
El grado de derrame pericárdico puede determinarse fácilmente por medio de ecocardiografía. Aunque el gasto cardiaco está reducido, rara vez se notan insuficiencia cardiaca congestiva y edema pulmonar. Hay controversias respecto a si el hipotiroidismo induce arteriopatía coronaria, pero la enfermedad de arteria coronaria es más común en pacientes con hipotiroidismo, lo cual quizá se relaciona con cifras aumentadas de colesterol total, colesterol de LDL, y posiblemente otros factores aterogénicos no tradicionales, como la lipoproteína A y la homocisteína. En pacientes con angina de pecho, el hipotiroidismo puede proteger el corazón contra el estrés isquémico, y la terapia de reemplazo puede agravar la angina al aumentar el consumo miocárdico de oxígeno.
2) deficiencia de hierro por aumento de la pérdida de dicho elemento con la menorragia, así como absorción intestinal alterada de hierro; 3) deficiencia de folato por alteración de la absorción intestinal de ácido fólico, y 4) anemia perniciosa, con anemia megaloblástica por deficiencia de vitamina B12. La anemia perniciosa a menudo forma parte de una agrupación de enfermedades autoinmunitarias, entre ellas hipotiroidismo debido a tiroiditis de Hashimoto o autoinmunitaria relacionada con autoanticuerpos contra la tiroides, anemia perniciosa vinculada con autoanticuerpos contra células parietales, diabetes mellitus relacionada con autoanticuerpos contra células de los islotes, e insuficiencia renal vinculada con autoanticuerpos contra las suprarrenales (síndrome de Schmidt). Estas enfermedades forman parte de lo que se ha denominado el síndrome de insuficiencia poliglandular.
Diagnóstico
La combinación de FT4 sérica baja y TSH sérica alta es diagnóstica de hipotiroidismo primario. Las cifras séricas de T3 son variables y pueden estar dentro del rango normal. Generalmente, la FT4 sérica es normal o normal baja, y la TSH sérica está un poco alta, situación que se denomina hipotiroidismo subclínico. Esto representa la forma más leve de hipotiroidismo, y es una consecuencia de la relación de la retroacción muy sensible entre la tiroides y la hipófisis.
En estas circunstancias, un pequeño decremento de la producción de hormona tiroidea por la tiroides, en el cual la concentración sérica de T4 aún está dentro del rango normal, suscita una concentración sérica de pH que está alta, aunque por lo general es de menos de 10 mU/L. El hipotiroidismo subclínico por lo general se debe a tiroiditis de Hashimoto subyacente, que puede confirmarse por medio de la evaluación de los títulos de anticuerpos anti-TPO. En pacientes con hipotiroidismo secundario o central, la FT4 sérica estará baja, y la TSH sérica estará baja o será normal, más que estar alta.
El paciente puede estar tomando hormona tiroidea (T4 en tabletas) cuando se le atiende por vez primera. Una glándula tiroides palpable o agrandada, y un resultado positivo en la prueba para autoanticuerpos contra la tiroides sugerirían tiroiditis de Hashimoto subyacente, en cuyo caso debe continuarse la administración del medicamento. En ausencia de anticuerpos antitiroideos, y si la indicación para terapia es incierta, el medicamento se debe suspender durante seis semanas y deben cuantificarse la FT4 y la TSH. El periodo de seis semanas de suspensión es necesario debido a la vida media prolongada de la T4 (siete días), y para permitir que la glándula hipófisis se recupere después de un periodo prolongado de supresión. En individuos hipotiroideos, la TSH sérica se torna alta a las 5 a 6 semanas y la FT4 permanece subnormal, mientras que ambas son normales después de seis semanas en individuos eutiroideos.
El cuadro clínico de mixedema desarrollado por completo generalmente es bastante claro, pero los síntomas y signos de hipotiroidismo leve o subclínico pueden ser muy sutiles o nulos. Esto ha llevado a la recomendación por algunas organizaciones profesionales de que se emprendan pruebas para detectar hipotiroidismo, especialmente en grupos de alto riesgo, como las mujeres de edad avanzada en las cuales la prevalencia es alta (hasta 20% en mujeres >65 años de edad) y en embarazadas, en las cuales el hipotiroidismo no tratado puede causar resultados adversos en el niño. A veces, los pacientes con hipotiroidismo se presentan con características poco comunes: neurastenia con síntomas de calambres, parestesias y debilidad musculares; anemia resistente a tratamiento; alteraciones de la función reproductora, incluso esterilidad, pubertad retrasada o precoz, o menorragia; edema idiopático o derrames pleuropericárdicos; retraso del crecimiento; constipación, rinitis o ronquera crónica debido a edema de la mucosa nasal o de las cuerdas vocales, y depresión grave que progresa hacia inestabilidad emocional o incluso psicosis paranoide manifiesta. En ancianos, el hipotiroidismo puede estar presente con apatía y retraimiento, que a menudo se atribuyen a senilidad. En esos casos, la evaluación de la función tiroidea con mediciones de la FT4 y la TSH séricas confirma o excluye hipotiroidismo como un factor precipitante.
Complicaciones
El paciente (o un miembro de la familia si el paciente está comatoso) puede recordar enfermedad tiroidea previa, tratamiento con yodo radiactivo o radioterapia en el área del cuello, o tiroidectomía.
El interrogatorio médico revela inicio gradual de letargo que progresa hacia estupor o coma. El examen revela bradicardia e hipotermia notoria, con temperatura corporal tan baja como de 24°C. El paciente por lo general es una anciana obesa con piel amarillenta, voz ronca, lengua grande, pelo delgado, ojos abotagados, íleo y reflejos lentos. Puede haber signos de otras enfermedades, como neumonía, infarto de miocardio, trombosis cerebral o sangrado gastrointestinal.
Pueden ocurrir crisis convulsivas, episodios de sangrado, hipocalcemia o hipercalcemia. Los indicios de laboratorio respecto al diagnóstico de coma mixedematoso son suero lactescente, caroteno sérico alto, colesterol sérico alto, y aumento de la proteína en el líquido cefalorraquídeo. Es posible que haya derrame pleural, pericárdico o abdominal con contenido alto de proteína. Las pruebas séricas revelan una FT4 baja y TSH notoriamente alta. Los autoanticuerpos contra la tiroides por lo general son fuertemente positivos, e indican tiroiditis de Hashimoto subyacente. El ECG muestra bradicardia sinusal y voltaje bajo. Si no hay estudios de laboratorio inmediatamente disponibles, lo cual suele suceder, el diagnóstico debe ser clínico.
La fisiopatología del coma mixedematoso comprende tres características principales: 1) retención de CO2 e hipoxia; 2) desequilibrio de líquido y electrólitos, y 3) hipotermia. La retención de CO2 y la hipoxia probablemente se deben en gran parte a depresión notoria de las respuestas ventilatorias a la hipoxia y la hipercapnia, aunque también pueden contribuir factores como obesidad, insuficiencia cardiaca, íleo, inmovilización, neumonía, derrame pleural o peritoneal, depresión del sistema nervioso central, y músculos débiles del tórax. El deterioro del impulso ventilatorio a menudo es grave, y en pacientes en coma mixedematoso casi siempre se necesita respiración asistida. La terapia con hormona tiroidea en pacientes con mixedema corrige la hipotermia y mejora la respuesta ventilatoria a la hipoxia. La intoxicación por agua, debida a perfusión renal disminuida y deterioro de la depuración de agua libre, es la principal alteración de líquidos y electrólitos. Esto causa hiponatremia y se maneja mejor mediante restricción de agua libre. La hipotermia a menudo no se reconoce, porque el termómetro clínico común sólo marca hasta alrededor de 34°C; es necesario usar un termómetro que registre una escala más amplia para obtener lecturas exactas de la temperatura corporal. El recalentamiento activo del cuerpo está contraindicado, porque puede inducir vasodilatación y colapso vascular. Un aumento de la temperatura corporal es una indicación útil de la eficacia de la terapia con T4. Los trastornos que pueden precipitar coma mixedematoso son insuficiencia cardiaca, neumonía, o administración de fármacos sedantes o narcóticos a un paciente con hipotiroidismo grave. La insuficiencia suprarrenal puede ocurrir ocasionalmente en relación con coma mixedematoso, sea debido a deterioro funcional de la hipófisis o a insuficiencia suprarrenal autoinmunitaria concurrente (síndrome de Schmidt). Se recomienda terapia concomitante con glucocorticoide hasta que pueda documentarse función suprarrenal normal mediante análisis de laboratorio. Tiene importancia diferenciar entre coma mixedematoso debido a insuficiencia primaria de la glándula tiroides, y el debido a insuficiencia hipofisaria (hipotiroidismo central). En esta última situación, es esencial el reemplazo de glucocorticoide. Los indicios clínicos respecto a la presencia de enfermedad hipofisaria son: un antecedente de amenorrea o impotencia, vello púbico o axilar escaso, y colesterol sérico normal, y cifras séricas normales o bajas de TSH. La CT o MRI puede revelar una silla turca agrandada.
La investigación de pacientes admitidos a servicios psiquiátricos, con FT4 y TSH séricas es una manera eficiente de identificar a estos pacientes, que suelen mostrar respuesta a la terapia con T4 sola o en combinación con agentes psicofarmacológicos. La eficacia de la terapia con T4 en pacientes hipotiroideos deprimidos ha dado lugar a la hipótesis de que la adición de T3 o T4 regímenes psicoterapéuticos para depresión quizá sea útil en pacientes sin enfermedad tiroidea demostrable, pero este concepto ha sido difícil de probar.
Tratamiento
Las dosis de reemplazo de T4 en adultos varía de 0.05 a 0.2 mg/día, con una media de alrededor de 0.125 mg/día. La dosis de T4 varía de acuerdo con la edad y el peso corporal del paciente. Debido al metabolismo de T4 más rápido, los lactantes y niños de corta edad requieren una dosis sorprendentemente alta de T4 en comparación con los adultos. En adultos, la dosis de reemplazo media de T4 es de aproximadamente 1.7 μg/kg/día. En adultos de edad avanzada, la dosis de reemplazo por lo general es más baja, de alrededor de 1.6 μg/kg/día. Para la supresión de TSH en pacientes después de tiroidectomía por cáncer tiroideo, la dosis promedio de T4 es de alrededor de 2.2 μg/kg/día. En la mayoría de los pacientes con hipotiroidismo, es posible empezar el tratamiento con el requerimiento de dosis estimado completo. Luego de 4 a 6 semanas, la dosis final se ajusta con base en la concentración sérica de TSH. El objetivo es normalizar la TSH sérica, lo cual típicamente es de 0.5 a 4 mU/L. En pacientes de edad avanzada o aquellos con enfermedad cardiaca subyacente, es mejor empezar con dosis bajas de T4 (p. ej., 0.025 mg al día), y aumentar la dosis a intervalos de 4 a 6 semanas con base en las mediciones de TSH sérica. Los estados de malabsorción, o la administración concurrente de productos de soya, antiácidos con hidróxido de aluminio, resinas de unión a ácidos biliares como la colestiramina y el colestipol, complementos de calcio, sucralfato, o compuestos de hierro, disminuyen la absorción de T4. En estos pacientes, la T4 debe administrarse antes del desayuno, cuando el estómago está vacío, y los otros compuestos deben tomarse 4 h más tarde. Otros fármacos, como el medicamento anticonvulsivo carbamazepina, pueden aumentar los requerimientos de hormona tiroidea al incrementar el metabolismo de T4. Los inhibidores de la bomba de protones, como el omeprazol, también pueden alterar la absorción de T4, posiblemente al disminuir el ácido gástrico, pero los datos al respecto son contradictorios. El reemplazo de estrógeno puede aumentar los requerimientos de T4 al incrementar la unión de T4 libre a la TBG, mientras que la terapia con andrógeno tiene el efecto opuesto. La T4 tiene una vida media suficientemente prolongada (siete días), de modo que si el paciente es incapaz de tomar medicamentos por vía oral durante algunos días, la omisión de la terapia con T4 no será perjudicial. Si un paciente hospitalizado se está manejando mediante terapia por vía parenteral sostenida, la dosis parenteral de T4 es de alrededor de 75 a 80% de la dosis por vía oral habitual.
Durante cierto tiempo, la tasa de mortalidad por coma mixedematoso fue de alrededor de 80%. El pronóstico ha mejorado tremendamente como resultado de reconocimiento de la importancia de la respiración mecánica asistida y el uso de T4 por vía intravenosa. En la actualidad el resultado probablemente depende de qué tan bien pueden manejarse las comorbilidades subyacentes y las consecuencias del hipotiroidismo grave.
En pacientes de edad avanzada, en particular aquellos con enfermedad cardiovascular conocida, es prudente empezar el tratamiento poco a poco. La T4 se administra en una dosificación de 0.025 mg/día durante 2 a 4 semanas; se aumentan 0.025 mg cada 2 a 4 semanas hasta alcanzar una dosis diaria de 0.075 mg; esta dosis se continúa durante alrededor de seis semanas. A continuación se mide la TSH sérica y se ajusta la dosificación en consecuencia. Típicamente se requieren de 2 a 4 meses para que un paciente llegue a equilibrio con dosificación completa. En algunos enfermos, el corazón es muy sensible a la concentración de T4 circulante, y si aparece angina de pecho o arritmia cardiaca, es esencial reducir la dosis de T4 de inmediato.
Efectos adversos de la terapia con T4
No hay casos reportados de alergia a la T4 pura, aunque es posible que un paciente presente una alergia al colorante o a algún componente de la tableta. Los principales efectos tóxicos de la sobredosificación de T4 son síntomas y signos de hipertiroidismo. Las palpitaciones son el síntoma cardiaco tirotóxico más común, y pueden ocurrir arritmias, en particular taquicardia o fibrilación auricular paroxística. El insomnio, el temblor, la inquietud y el calor excesivo también pueden ser problemáticos. El simple hecho de omitir las dosis diarias de T4 durante tres días y después reducir la dosificación corregirá el problema.
La resorción ósea aumentada y la osteoporosis se han relacionado con hipertiroidismo de larga evolución, y aparecerán también en mujeres posmenopáusicas tratadas en exceso de manera crónica con T4. Esto puede evitarse mediante vigilancia regular y al mantener concentraciones séricas normales de TSH en pacientes que están recibiendo terapia de reemplazo a largo plazo. En sujetos que reciben terapia supresora de TSH para cáncer tiroideo, si la concentración sérica de FT4 se mantiene dentro del límite normal superior, incluso si la TSH está suprimida, los efectos adversos de la terapia con T4 sobre los huesos serán mínimos. Además, la administración concomitante de estrógeno o bifosfonatos a mujeres posmenopáusicas que están recibiendo terapia con T4 en dosis altas, minimiza la resorción ósea.
Evolución y pronóstico
La evolución del hipotiroidismo no tratado consiste en deterioro lento, lo que en potencia lleva finalmente a coma mixedematoso y muerte. Aun así, con tratamiento apropiado, el pronóstico a largo plazo es excelente. Como consecuencia de la vida media prolongada (siete días) de la T4, se requiere tiempo para establecer equilibrio en una dosis fija. Por lo tanto, tiene importancia vigilar la FT4 y la TSH séricas cada 4 a 6 semanas hasta que se alcance un equilibrio. A partir de entonces, la FT4 y la TSH pueden vigilarse una vez cada 6 a 12 meses. La dosis de T4 se aumenta alrededor de 50% en la mayoría de las mujeres durante el embarazo; los mayores incrementos de la dosificación se realizan en mujeres que tienen menor reserva tiroidea (esto es, en mujeres en quienes se ha practicado una tiroidectomía o que han recibido terapia con yodo radiactivo). Los pacientes de edad avanzada metabolizan la T4 más lentamente, y es probable que la dosis tenga que disminuirse de manera gradual con la edad.
La tirotoxicosis es el síndrome clínico que se produce cuando los tejidos quedan expuestos a concentraciones altas de hormonas tiroideas circulantes. Da por resultado aceleración generalizada de procesos metabólicos. La tirotoxicosis casi siempre se debe a hiperactividad de la glándula tiroides, o hipertiroidismo. En ocasiones, la tirotoxicosis puede debe
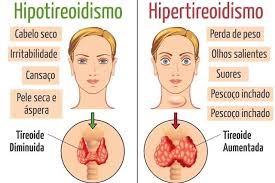
rse a otras causas, como ingestión excesiva de hormona tiroidea o, muy rara vez, secreción excesiva de hormonas tiroideas a partir de un tumor ovárico (estruma ovárico).
(ENFERMEDAD DE GRAVES)
La enfermedad de Graves es la forma más común de tirotoxicosis. Las mujeres quedan afectadas alrededor de cinco veces más comúnmente que los varones. La enfermedad puede ocurrir a cualquier edad, con una incidencia máxima en el grupo de 20 a 40 años. El síndrome consta de uno o más de los datos que siguen: 1) tirotoxicosis; 2) bocio; 3) oftalmopatía (exoftalmos), y 4) dermopatía (mixedema pretibial).
Etiología
La enfermedad de Graves en la actualidad se considera una enfermedad autoinmunitaria de origen desconocido. Hay una fuerte predisposición familiar, alrededor de 15% de los pacientes con enfermedad de Graves tiene un familiar cercano con el mismo trastorno, y aproximadamente 50% de los familiares de pacientes con enfermedad de Graves tiene autoanticuerpos circulantes contra la tiroides. Hay una concordancia mucho más alta de enfermedad de Graves en gemelos monocigóticos en comparación con la que se observa en gemelos dicigóticos, y puesto que la tasa de concordancia en gemelos monocigóticos es de mucho menos de 100%, debe haber factores ambientales que también participan. Los desencadenantes ambientales propuestos comprenden estrés, consumo de tabaco, infección y exposición a yodo. El estado posparto, que puede relacionarse con aumento de la función inmunitaria, también puede desencadenar enfermedad de Graves en mujeres susceptibles desde el punto de vista genético.
Patogenia
En la enfermedad de Graves, los linfocitos T quedan sensibilizados a antígenos dentro de la glándula tiroides, y estimulan linfocitos B para que sinteticen anticuerpos contra estos antígenos.
Un anticuerpo de ese tipo se dirige contra el receptor de TSH en la membrana celular tiroidea, lo que estimula el crecimiento de la glándula tiroides y la función de la misma. El anticuerpo se llama anticuerpo estimulante de la tiroides (TSAb), o inmunoglobulina estimulante de la tiroides (TSI). La presencia de este anticuerpo circulante guarda correlación positiva con enfermedad activa y con recaída de la enfermedad después de terapia con fármacos antitiroideos. Hay una predisposición genética subyacente, pero no está claro qué desencadena el comienzo inicial del hipertiroidismo. Algunos factores que pueden incitar la respuesta inmunitaria propia de la enfermedad de Graves son: 1) embarazo, en particular el periodo posparto; 2) el exceso de yoduro, particularmente en áreas geográficas en las cuales hay deficiencia de yoduro, donde la falta de este último puede mantener a raya una enfermedad de Graves latente; 3) interferón alfa, quizás al modificar la capacidad de respuesta inmunitaria; 4) infecciones virales o bacterianas, y 5) estrés psicológico. La patogenia de la oftalmopatía puede comprender linfocitos citotóxicos (células asesinas) y anticuerpos citotóxicos sensibilizados a un antígeno común como el receptor de TSH que se encuentra en fibroblastos orbitarios, músculo orbitario y tejido tiroideo. Las citocinas provenientes de estos linfocitos sensibilizados pueden causar activación y proliferación de fibroblastos y preadipocitos orbitarios, lo que da como resultado aumento de las cantidades de grasa y glucosaminoglucanos retroorbitarios, así como tumefacción de los músculos extraoculares, produciendo proptosis (protrusión) de los globos oculares y diplopía, así como enrojecimiento, congestión y edema conjuntival y periorbitario (oftalmopatía u orbitopatía relacionada con la tiroides). Por razones que no están claras, el tabaquismo es un potente factor de riesgo para la aparición de oftalmopatía relacionada con la tiroides. La patogenia de la dermopatía tiroidea (mixedema pretibial) y la rara inflamación subperióstica en las falanges de las manos y los pies (osteopatía o acropaquia tiroidea) también pueden comprender la estimulación de fibroblastos en estas ubicaciones por citocinas de linfocitos. Muchos síntomas de tirotoxicosis sugieren un estado de exceso de catecolamina, incluso taquicardia, temblor, sudoración, asinergia oculopalpebral y mirada fija, sin embargo, las concentraciones circulantes de adrenalina y noradrenalina son normales o bajas; así, en la enfermedad de Graves el cuerpo parece tener sensibilidad aumentada a las catecolaminas. Esto puede deberse en parte a incremento (mediado por hormona tiroidea) de receptores beta-adrenérgicos unidos a membrana en diversos tejidos.
Datos clínicos
Werner ha clasificado los signos oculares de la enfermedad de; esta clasificación es útil para describir la extensión de la afección ocular. Empero, no es útil para vigilar el proceso de la enfermedad, porque una clase no necesariamente progresa hacia la siguiente. La clase 1 comprende retracción de los párpados superiores relacionada con tirotoxicosis activa y por lo general se resuelve de manera espontánea cuando la tirotoxicosis se controla satisfactoriamente. La retracción de los párpados puede observarse en cualquier forma de tirotoxicosis, porque se debe a estimulación adrenérgica del párpado superior. Las clases 2 a 6 representan enfermedad infiltrativa verdadera que afecta los músculos y tejidos orbitarios, y son específicas para enfermedad de Graves. La clase 2 se caracteriza por afección de tejido blando con edema periorbitario, congestión o enrojecimiento, y edema de la conjuntiva (quemosis). La clase 3 consta de proptosis según se mide con el exoftalmómetro de Hertel; este instrumento consta de dos prismas con una escala montada en una varilla. Los prismas se colocan en los rebordes orbitarios laterales, y la distancia desde el reborde orbitario hasta la parte anterior de la córnea se mide en la escala. La clase 4 consta de afección de músculos extraoculares, que típicamente se debe a fibrosis y falta de relajación muscular, lo que limita la función del músculo antagonista. El recto inferior es el músculo más comúnmente afectado en el proceso infiltrativo. Al no relajarse normalmente, la limitación de la mirada hacia arriba es el signo más común en pacientes con afección de músculos oculares. El siguiente músculo más comúnmente afectado es el recto medial, lo que altera la mirada lateral. La clase 5 se caracteriza por afección corneal (queratitis) debida a incapacidad para cerrar los ojos por completo. La clase 6 es una pérdida de la visión por afección del nervio óptico, probablemente debido a isquemia del nervio por compresión de los músculos extraoculares agrandados circundantes. Como se mencionó, la oftalmopatía tiroidea se debe a infiltración de fibroblastos orbitarios y de los músculos extraoculares con linfocitos, y líquido de edema debido a una reacción inflamatoria. La órbita es un cono encerrado por hueso, y la tumefacción de los músculos extraoculares dentro de este espacio cerrado causa proptosis del globo y alteración del movimiento muscular, lo que da por resultado diplopía. El agrandamiento del músculo ocular puede demostrarse mediante ecografía o CT orbitaria, o MRI. Si bien sólo alrededor de una tercera parte de los pacientes tiene afección ocular en clínica, en más de 90% pueden detectarse músculos agrandados mediante estudios de imágenes. La dermopatía tiroidea consta de engrosamiento de la piel, en particular sobre la parte baja de la tibia, debido a acumulación de
glucosaminoglucanos. Es relativamente rara; ocurre en alrededor de 2 a 3% de los pacientes con enfermedad de Graves. Por lo general se relaciona con oftalmopatía importante y con un título sérico muy alto de TSAb. La piel está notoriamente engrosada, con una superficie en cáscara de naranja, y no puede tomarse entre los dedos. A veces la dermopatía afecta toda la pierna, y puede extenderse hasta los pies. Finalmente, la afección ósea (osteopatía o acropaquia tiroidea), con formación de hueso subperióstico e hinchazón, es en particular evidente en los huesos metacarpianos como dedos de las manos hipocráticos, este también es un dato relativamente raro. Un dato más común en la enfermedad de Graves es la separación entre las uñas y los lechos ungueales de las manos (onicólisis o uñas de Plummer), probablemente causado por crecimiento rápido de las uñas.
1. Datos de laboratorio
La combinación de una FT4 alta y TSH suprimida hace el diagnóstico de hipertiroidismo. Un 5% de los enfermos tiene concentración normal de FT4, pero alta de T3 sérica, una situación que se denomina tirotoxicosis por T3. Asimismo, la enfermedad de Graves muy leve puede dar por resultado concentraciones séricas de FT4 y T3 que están dentro del rango normal pero que están suficientemente altas como para llevar a supresión de la concentración sérica de TSH, una situación denominada hipertiroidismo subclínico. Si hay aumento tanto de la FT4 como de la TSH, y la RAIU también está alta, se considera la presencia de un tumor hipofisario secretor de TSH o síndromes de resistencia generalizada o hipofisario. Los pacientes muy graves pueden tener concentración sérica baja de TSH, pero también tienen cifras séricas bajas de FT4 y T3 (el llamado síndrome del eutiroideo enfermo). En la enfermedad llamada hipertiroxinemia disalbuminémica familiar, hay una albúmina anormal en el suero, que se une de preferencia a T4 pero no a T3. Esto da lugar a aumento de la T4 y de FT4I séricas, y a menudo de la T4 libre cuando se mide mediante ensayos de T4 libre análogos clínicos habituales, pero la T4 libre por lo general es normal mediante métodos de diálisis de equilibrio, al igual que la T3 y la TSH séricas. Es importante diferenciar entre este estado eutiroideo e hipertiroidismo verdadero. Además de la ausencia de datos clínicos de hipertiroidismo, una concentración sérica normal de T3 y normal de TSH excluye hipertiroidismo. Si hay signos oculares, el diagnóstico de enfermedad de Graves puede hacerse sin más análisis. En ausencia de signos oculares, y si el paciente es hipertiroideo con bocio o sin él, debe efectuarse una prueba de RAIU. Una captación alta es típica de enfermedad de Graves.
En contraste, se observa captación baja en pacientes con varias formas de hipertiroidismo que se resuelve de manera espontánea, como en la tiroiditis subaguda o la tiroiditis silenciosa también llamada tiroiditis posparto. También se encuentra captación baja de yodo radiactivo en pacientes que reciben una carga de yodo o que están en terapia con T4 o, rara vez, en asociación con estruma ovárico en el cual hay tejido tiroideo ectópico dentro de un teratoma ovárico. Los autoanticuerpos contra la tiroides contra tiroglobulina y TPO por lo general están presentes en el suero tanto en la enfermedad de Graves como en la tiroiditis silenciosa, una variante de la tiroiditis de Hashimoto, pero los antiTSH son relativamente específicos para la enfermedad de Graves. Esto puede ser una prueba diagnóstica útil en el paciente que se presenta con exoftalmos unilateral o bilateral sin signos obvios o manifestaciones de laboratorio de enfermedad de Graves (la llamada “enfermedad de Graves eutiroidea”). La gammagrafía con 123 I o con tecnecio es útil para evaluar el tamaño de la glándula, así como la presencia de nódulos calientes o fríos. La CT y la MRI de las órbitas revelan agrandamiento de los músculos extraoculares en la mayoría de los pacientes con enfermedad de Graves, incluso cuando no hay evidencia clínica de oftalmopatía. En pacientes con oftalmopatía clínica, el agrandamiento de los músculos orbitarios puede ser notorio.
Complicaciones
La crisis tirotóxica (tormenta tiroidea) es la exacerbación aguda de todos los síntomas y signos de tirotoxicosis; a menudo se presenta como un síndrome cuya gravedad puede poner en peligro la vida. En ocasiones, la tormenta tiroidea puede ser leve y presentarse simplemente como una reacción febril inexplicable después de intervención quirúrgica tiroidea en un paciente que se ha preparado de manera inadecuada. Más comúnmente, ocurre en una forma más grave después de una operación, terapia con yodo radiactivo o parto, en un paciente con tirotoxicosis controlada de manera insatisfactoria, o durante una enfermedad o trastorno grave y estresante, como diabetes no controlada, traumatismo, infección aguda, reacción grave a fármacos, o infarto de miocardio. Las manifestaciones clínicas de la tormenta tiroidea son hipermetabolismo notorio y respuesta adrenérgica excesiva. La fiebre varía de 38 a 41°C, y se relaciona con rubor y sudoración. Hay taquicardia notoria, a menudo con fibrilación auricular y presión del pulso alta; en ocasiones ocurre insuficiencia cardiaca. Los síntomas del sistema nervioso central son agitación, inquietud, delirio notorios y coma. Los síntomas gastrointestinales son náusea, vómito, diarrea e ictericia. Un resultado mortal se relaciona con insuficiencia cardiaca y choque. En una época se creyó que la tormenta tiroidea se debía a liberación o descarga repentina de T4 y T3 almacenadas, desde la glándula tirotóxica. Empero, estudios cuidadosos han revelado que las concentraciones séricas de T4 y T3 en pacientes con tormenta tiroidea no necesariamente son más altas que en pacientes tirotóxicos sin esta afección. No hay evidencia de que la tormenta tiroidea se deba a producción excesiva de T3. Asimismo, en la tirotoxicosis, el número de sitios de unión adrenérgicos para catecolaminas aumenta, de modo que los tejidos cardiaco y nervioso tienen aumento de la sensibilidad a catecolaminas circulantes. Además, hay unión disminuida a TBG con aumento adicional de la T3 y T4 libres. Una teoría es que en estas circunstancias, con sitios de unión aumentados disponibles para catecolaminas, una enfermedad, infección o estrés quirúrgico agudo desencadenan un torrente de catecolaminas que, en asociación con cifras altas de T4 y T3 libres, precipita el problema agudo. La característica diagnóstica clínica más notoria de la crisis tirotóxica es la hiperpirexia fuera de proporción con otros datos. Los datos de laboratorio son T4, FT4 y T3 séricas altas, así como TSH suprimida.
Tratamiento de la enfermedad de Graves
Aunque el síndrome de enfermedad de Graves depende de mecanismos autoinmunitarios, el manejo se ha dirigido en su mayor parte hacia el control del hipertiroidismo. Se dispone de tres métodos buenos: farmacoterapia antitiroidea; 2) intervención quirúrgica, y 3) terapia con yodo radiactivo.
50% de los pacientes, pero puede no durar toda la vida. Por lo general se inicia farmacoterapia antitiroidea con dosis más grandes. Cuando el paciente se hace eutiroideo desde el punto de vista bioquímico después de 4 a 12 semanas, puede lograrse terapia de mantenimiento con una dosis más baja. Debido al potencial de hepatotoxicidad grave y en potencia mortal con PTU, el metimazol es el mejor fármaco antitiroideo para la mayoría de los pacientes. El metimazol tiene una duración de acción más prolongada, y la dosificación diaria única lleva a mejoría del apego a las indicaciones. Un régimen típico empieza con una dosis de 10 a 20 mg de metimazol cada mañana durante
1 a 2 meses; estas dosis a continuación se reducirían a 5 a 10 mg cada mañana para terapia de mantenimiento. El PTU podría considerarse en pacientes con reacciones alérgicas leves al metimazol, y se prefiere en embarazadas durante el primer trimestre debido a raros efectos teratogénicos del metimazol en el feto. Un régimen de uso común consta de administrar PTU, 100 mg cada 8 h inicialmente, y después en 4 a 8 semanas, con reducción de la dosis a 50 a 200 mg 1 o 2 veces al día. Las pruebas de laboratorio más valiosas en la vigilancia temprana de la evolución de la terapia son la FT4 y T3 séricas. La concentración de TSH a menudo permanece suprimida durante muchas semanas o incluso meses; por ende, no es un índice fiable de la función tiroidea al principio del tratamiento. En un método de terapia alternativo se utiliza el concepto de un bloqueo total de la actividad tiroidea. Se trata al paciente con metimazol hasta que es eutiroideo (durante alrededor de 3 a 6 meses), pero en lugar de seguir disminuyendo paulatinamente la dosis de fármaco antitiroideo, en este punto se agrega T4 en una dosis de alrededor de 0.1 mg/día. El paciente a continuación sigue recibiendo la combinación de fármaco antitiroideo y T4 (0.1 mg/día) durante otros 12 a 24 meses. Al final de este tiempo, o cuando el tamaño de la glándula ha vuelto a lo normal, se suspenden los fármacos. Esta terapia combinada evita la aparición de hipotiroidismo debido a dosis excesivas de fármacos antitiroideos, pero la frecuencia de recaída es casi la misma que después de tratamiento con fármacos antitiroideos solos. Es más caro, porque se necesitan dos medicamentos, y puede haber mayor frecuencia de efectos secundarios. Por ende, esta estrategia no se recomienda para la mayoría de los pacientes.
La duración de la terapia con fármacos antitiroideos en la enfermedad de Graves por lo general es de 12 a 24 meses; algunos pacientes prefieren recibir tratamiento, si es necesario, durante periodos prolongados (p. ej., decenios), pero esto no es típico. Una remisión sostenida es más probable en las circunstancias que siguen: 1) si la glándula tiroides vuelve al tamaño normal; 2) si la enfermedad puede controlarse con una dosis relativamente pequeña de fármacos antitiroideos, y 3) si los TSAb ya no son detectables en el suero al final del periodo de terapia.
PTU también puede preferirse en pacientes con tormenta tiroidea, en los cuales la inhibición de la conversión de T4 en T3 puede ser importante en clínica.
El hipotiroidismo también puede ocurrir como un suceso tardío después de un periodo de farmacoterapia antitiroidea más temprana para enfermedad de Graves; en esos pacientes, la enfermedad de Graves consumida probablemente es el resultado final de destrucción autoinmunitaria de la tiroides. En consecuencia, todos los pacientes con enfermedad de Graves que no se han hecho hipotiroideos requieren vigilancia de por vida a fin de asegurarse de que permanezcan eutiroideos.
Elección de la terapia
La elección de la terapia varía con la naturaleza y la gravedad de la enfermedad y con las costumbres prevalentes; por ejemplo, en Estados Unidos, la terapia con yodo radiactivo ha sido el tratamiento preferido para el paciente promedio, mientras que en Europa y Asia se prefiere la farmacoterapia antitiroidea primaria. En general, los fármacos antitiroideos son una terapia inicial razonable para niños y adolescentes, y en adultos con enfermedad leve y bocios pequeños. En todos los otros pacientes el yodo radiactivo es el mejor tratamiento. En la actualidad, la participación de la intervención quirúrgica se limita a pacientes que no se apegan a las indicaciones y que rehúsan el yodo radiactivo, pacientes con un bocio grande, y otras circunstancias poco comunes antes mencionadas.
Tratamiento de complicaciones
Los estudios de laboratorio revelan FT4I y o FT4 alta, aumento notorio de T3, y por lo general TSH baja, en contraste con los lactantes normales, que tienen TSH alta en el momento del nacimiento. La edad ósea puede estar acelerada; por lo general se encuentran TSAb en el suero tanto del lactante como de la madre. Se cree que la patogenia de este síndrome comprende transferencia transplacentaria de TSAb desde la madre hacia el feto, con aparición subsiguiente de tirotoxicosis. La enfermedad es autolimitada y disminuye durante un periodo de 4 a 12 semanas, lo cual coincide con el descenso de la concentración sérica de TSAb en el niño. La terapia para el lactante comprende PTU en una dosis de 5 a 10 mg/kg/día (en dosis divididas a intervalos de 8 h); solución de Lugol, una gota (8 mg de yoduro de potasio) cada 8 h, y propranolol, 2 mg/kg/día en dosis divididas. Además, están indicada nutrición adecuada, antibióticos para infección si la hay, sedantes si es necesario, y terapia de sostén. Si el niño tiene tirotoxicidad acentuada, la terapia con corticosteroide (prednisona, 2 mg/kg/día) bloquea parcialmente la conversión de T4 en T3, y puede ser útil durante la fase aguda. Los medicamentos anteriores se reducen de manera gradual a medida que el niño mejora, y por lo general pueden suspenderse hacia las 6 a 12 semanas. Los sueros maternos también pueden contener TSAb que funcionan como anticuerpos bloqueadores que pueden cruzar la placenta y producir hipotiroidismo transitorio en el lactante. Quizá sea necesario tratar esta afección mediante complementos de T4 durante un tiempo breve.
– Patología frecuente 1/1000000, desde poco hasta muy agresivo.
– Problemas fundamentales que plantean para tomar las decisiones: Difícil diagnostico,
Edad, Tipo histológico sospechado, Tamaño del tumor, Invasión linfática, Factores que modifican el pronóstico.
– Tipo histológico: Papilar, casi benigna, Folicular, Mixto papilar y folicular, Medular
Anaplasico: el mas agresivo de todos, Linfoma. Esto ha cambiado con los nuevos tratamientos de quimioterapia y radioterapia.
– Edad: A mas edad mas mortalidad. Adulto joven: 50% nódulos son malignos pero con buena evolución. Edad mayor de 45 años aumenta mucho la mortalidad.
– Tamaño y extensión linfática: < 1.5 cm> , sin invasión linfática suelen ser benignos. Y si hay Afectación linfática: empeora el pronostico.
EN UN ESTUDIO ANTEROPOSTERIOR:
Adenocarcinoma:
Cáncer papilar:
El mas benigno y ademas el mas frecuente (50-80%), es un tumor bien diferenciado, mas frecuente en mujeres a edad joven (20-40), es el mas silente clínicamente ( esto no están cierto porque el folicular que es un poco mas agresivo también es muy silente). Es un tumor pequeño, menor de 1 cm, pero que puede ser multicentrico, esto nos puede obligar a resecar el tiroides contralateral (pensamos que el otro lado puede tener un microcarcinoma).
Variante: papilarfolicular, mezcla de elementos celulares Es un tumor bien diferenciado, presenta psamomas o áreas de calcificación, característico de este tipo. Hay afectación linfática subclínica, microafectación en el 50% de los casos, estos ganglios no serán palpables, esto quiere decir que hay que hacer biopsia de los ganglios linfáticos para ver si hay afectación.
Folicular:
Predominio en zonas con déficit de yodo, mujeres jóvenes (2:4), escasa clínica. Es un tumor algo mas grande, es encapsulado. La diferencia mas importante con respecto al papilar, es que este no tiene tanta tendencia a infiltrar los linfáticos (10%). Enfocamos la tiroidectomia, pero no se busca con tanto ímpetu los ganglios, solo el primer ganglio , pero debemos de tener en cuenta que suele dar con mas frecuencia metástasis a distancia ( óseas y pulmonares). ( al contrario que el papilar que suele dar mas metástasis linfáticas y recidivas locales cuando son mas agresivos, el folicular es mas hematógeno, metástasis a distancia). Es un tumor muy vascularizado, encapsulado y funcionante, esto es bueno porque podemos tratar las metástasis a distancia con yodo radiactivo. Constituido por folículos que invaden el parénquima. Es una variedad que aumento mucho tras la catástrofe de Chernobil, en niños, con la variedad solida folicular, con una serie de alteraciones en la rotura del ADN e inversión del cromosoma 10, por lo que es un efecto carcinogenico que afecta mas a niños. La radiación por tanto aumenta el riesgo de tumor tiroideo en una proporción mayor en niños.
Anáplasico:
Poco frecuente 10%, en mujeres a edad avanzada, en zonas de bocio endémico, en estos grandes bocios se desarrolla a la larga un indiferenciacion celular que da el carcinoma anáplasico. Es un tumor no funcionante, con células indiferenciadas, capacidad de crecimiento muy elevada, infiltra capsula, linfáticos, tráquea y músculos del cuello, por lo que por su agresividad local da síndrome asfíctico y muerte en cuestión de meses. Suele tener metástasis linfáticos. Podemos tener incluso ulceraciones cutáneas.
Carcinoma de células de Hürtle:
Puede ser un adenoma o carcinoma que se diferencia según el tipo de células, es poco
frecuente. En ocasiones se da con una tiroiditis de Hashimoto. La diferencia entre adenoma y carcinoma depende de la histología y del comportamiento, este es un carcinoma donde hay que hacer tiroidectomia y linfadenectomia, porque puede tener un comportamiento muy agresivo.
Epidermoide: Muy poco frecuente, Medular: 3-10%, Tumor de celular parafoliculares. Linfomas y sarcomas: muy raros.
ESTUDIO CLINICO Y DIAGNOSTICO:
Sintomatología: – Prácticamente nula, en ocasiones se da en enfermos con hipertiroidismo, pero no es muy frecuente. Lo canceres de tiroides se diagnostican porque tienen nódulos no por hipertiroidismo. En casos de hipotiroidismo podemos encontrar cáncer de tiroides, pero no esta en relación con la función.
– Por lo que no hay relación con cuadros distiroideos
– Lo que vemos es una tumoración, no hay expresividad en técnicas de laboratorio
– Se diagnostica con ECO y PAAF.
Pero tener en cuenta:
– Edad y sexo del paciente ( nódulo en joven , sospechar malignidad), es mas frecuente en la mujer, pero un nódulo en el hombre tiene mayor probabilidad de cáncer.
– Historia de radiación previa
– Tumor solido o quístico
Evaluación diagnostica:
– Exploración: tamaño, consistencia, adherencia a piel o ECM, piel( le pedimos que degluta para ver si se mueve la glandula- duro, que no se moviliza, aumentado de
tamaño, pensaremos en tumor)
– Cadenas ganglionares , cuando palpamos adenopatías, tendremos mas sospecha de
que se trate de un tumor. Salvo tiroiditis tipo hashimoto, no hay adenopatías.
– Exploración general del enfermos: no aporta datos de interés.
– ECO: fácil de realizar, no es agresivo. Nos aporta densidad, limites, afectación ganglionar, nos da la heterogeniedad dentro del tumor (solido, liquido, calcificaiones o psamomas ( papilar)…), nos permite ademas hacer una PAAF.
– Eco doppler: con esto podemos ver la vascularización, que a mas intensa mas
probabilidad e folicular.
– Gammagrafía tiroidea: hoy día se hace menos, con los eco de última generación se ven
muy bien las características de las lesiones, y tenemos ecografistas especialistas y que
hacen buenas punciones. – TAC tiroideo: no se usa como diagnóstico para ver que tipo histológico es, solo nos sirve para ver si hay infiltración fuera de la glándula: adenopatías o lesiones a distancia
– Estudio de extensión: Rx torax, Gammagrafía ósea
TRATAMIENTO: En los tumores diferenciados: nodulectomía o hemitiroidectomía puesto que son benignos en su evolución ( tumores menores de 2 cm, pacientes menores de 40 años) , las guías clínicas modernas están diciendo que lo mejor es hacer tiroidectomia. Este cambio se debe a que el entrenamiento quirúrgico disminuye la morbimortalidad de la tiroidectomia, antes se temían los problemas de hipoparatiroidismo por llevarse la paratiroides, lesiones del recurrente. Tiroidectomia total o subtotal, esto da también ventaja para hacer los rastreos de metástasis postquirurgicas, porque lo que queda de glándula captarían el contraste.
También con la tiroidectomia total o subtotal: Evitamos las recidivas contralaterales, – Tendencia a trasformación anaplásica en anciano, No evita la necesidad de hormonoterapia ( esto es algo imprevisible, con hemitiroidectomia no precisamos en ocasiones de hormonoterapia, pero otros si, por lo que siempre debemos de hacer control analitico).
– Si hay que tratar con I131 (mtt): menor dosis, si hay remanaente tiroideo va a captar
todo el yodo, por lo que el tratamiento será menos eficaz.
– Tiroglobulina marcador de tumor debe desaparecer tras tiroidectomia. Con la tiroidectomia total si aparecen niveles de tiroglobulina nos podria servir como marcador de tumor.
Linfadenectomia: no se realiza de manera profiláctica, vaciamiento ganglionar en presencia de afectación, se realiza mediante biopsia intraoperatoria.
Sistemáticamente en el papilar, haremos un vaciamiento de los ganglios que hay debajo del itsmo, si vemos que hay afectación de los ganglios, haremos una linfadenectomia del lado homolateral completa. En el folicular no es un acto sistematico a no ser que haya afectación.
FACTORES DE RIESGO DE MALIGNIDAD DE NODULOS:
– Nódulo solitario, duro, fijo. ( folicular, anaplásico, carcinoma medular, o anaplásico)
– Crecimiento rápido ( medular o anaplásico, mas malignos)
– Alteraciones de la voz ( infiltración del recurrente, algo crece deprisa)
– Presencia de linfadenopatías
– No disminuye con el tratamiento ( distiroidectomias, suelen tener buena respuesta al
tratamiento) ( ejemplo, bocio que no responde a tratamiento, puede ser que se a
autónomo o que sea un tumor)
– Gammagraficamente frio ( mayor indicación de malignidad, los calientes raramente
son malignos)
– Paciente joven ( mas tendencia a que sea un carcinoma)
– Nódulo solido en la ecografía ( más frecuente que liquido de ser maligno)
– Historia de: radiación (riesgo de carcinoma mayor), o de neoplasia tiroidea familiar,
neoplasia pluriendocrina familiar.
Eco-PAAF:
La usamos para tomar una decisión quirúrgica, hay casos que no tenemos tan claro que
realizar tras el resultado de la PAAF. Las lesiones quísticas más frecuentemente son benignas, pero podemos tener un falso negativo. PAAF que nos da benignidad y no cumple criterios de sospecha de neoplasia adoptaremos una actitud conservadora.
Lesión sólida y la PAAF dice que es maligna, opera. Otras posibilidad es que la PAAF no este clara, diga que es una proliferación altamente celular, si a esto añadimos criterios de malignidad por parte del enfermo, en estos casos plantearemos una indicación quirúrgica, al igual que con los de las células de Hürtle que pueden ser malignos.
Carcinoma medular: Es tan diferente en su celularidad y comportamiento que debemos de estudiarlo de manera separada. 1958 Hazard, Crile: descripción del Ca medular. Clínica y histologíacamente diferente del anaplasico 1961 Sippele: observa un aumento de la incidencia del cáncer de tiroides, en pacientes con feocromocitomas 1962 Copp: calcitonina ( polipeptido hipocalcemiante) hallazgo de la misma en la tiroides.
Carcinoma medular derivado de las células C productoras de calcitonina, tienen una actividad secretora muy intensa, no solo va a segregar calcitonina.
Se asocia a otros trastornos endocrinos y puede ser:
– Esporádico: 85-90% de los casos, bilateral 15%
– Familiar 10-15% de los casos ( hasta 25%), gen autosómico dominante ( penetración variable), bilateral en el 80% de los familiares. Contexto de neoplasia endocrina multiple
ETIOPATOGENIA:
Se ha encontrado una mutacion en el ADN de las células tumorales del CMT, relaciones con el protooncogen RET. ( CR10). Se expresaría una enzima mutada de las tirosincinasas RET. Esta implicada en la regulación del crecimiento y diferenciación celular de todos los CMT. En un paciente con un posible carcinoma medular, determinamos el gen en el enfermo y si esto es +, haremos las determinaciones en toda la familiar. El tumor produce calcitonina y CEA, pero se sospecha que puede haber tumores incipientes que no dan elevación de calcitonina, por lo que esta es fundamental en el enfermo que
sabemos que esta afectado, porque nos sirve de marcador de tumor y de ver si hemos extirpado el tumor y sus metástasis o recidivas si la monitorizamos en el tiempo. En la familia podemos hacer determinaciones de calcitonina, pero con una enfermedad incipiente podemos no encontrar valores alterados, ademas este seguimiento es costoso, por lo que haremos la determinación del gen RET si el paciente enfermo tiene la mutacion y esto nos a va llevar a que todos los familiares con la alteración del gen RET, deben de ser sometidos a una TIROIDECTOMIA PROFILACTICA. La herencia de esta mutacion es autosómica dominante.
ANATOMIA PATOLOGICA: Crecimiento nodular muy duro, constituido por células parafoliculares con grandes núcleos hipercromicos.
ACTIVIDAD SECRETORA:
– Calcitonina: tets diagnostico en familiares, seguimiento postoperatorio, detectar
metástasis.
– Histaminasa
– Pgs
– Serotonina
– Dopa descarboxilasa
– Beta endorfina
– Factor de crecimiento nerviosos
– Somatostatina
– ACTH y CRH
SINTOMATOLOGIA Y DIAGNOSTICO:
Sospecha diagnostica en pacientes con patología tiroidea asociada a hipocalcemia, la patología del tiroides benigna o maligna no suele alterar la calcemia, pero si tenemos alteración de esta debemos de sospechar un aumento de actividad de la calcitonina.
Igual en ambos sexos. Aparición de un bocio duro ( nódulo)
Datos obligados de anamnesis: sudoración, cefalea, diarrea ( 30%), enrojecimiento, antecedentes familiares, HTA,.. debido al resto de hormonas que produce. Esta sintomatología no se parece a la de un enfermos con un hipo/hipertiroidismo. ( no hay tanto temblor ni sudor de las manos ni alteración ocular como habría por ejemplo en un basedow)
DIAGNOSTICO:
– Estudio funcional tiroideo: normal
– Pruebas de estimulo y creación: norma
– Gammagrafía tiroidea: nódulo frio
– Eco: nódulo solido
– Rx cervical: calcificaiaciones en la región tiroidea, estas pueden ser tan importantes
que se ven en una simple Rx
– Hecho patognomónico. Elevación de calcitonina mayor de 0.2 ng/ml
– Aumento de CEA
– Aumento de histaminasa en sangre
– Es obligado buscar la enfermedad pluriendocrina: estudio de suprarrenal (
feocromocitoma), buscar alteraciones de la paratohormona, Rm abd para ver
alteraciones pancreáticas…
ESTADIAJE
Buscamos la región cervical, lesiones en toras, lesiones hepáticas u óseas. Hacemos un estudio de la familia sobretodo en el medular bilateral ( pero en principio siempre)
Determinación gen RET al enfermo.
TRATAMIENTO:
Evolucion natural es peor que el papilar o folicular ( mejor que anaplasico)
Es quirúrgico siempre, son células que no captan yodo.
Empeoran el pronóstico:
– Edad, peor 30-40 años
– Invasión ganglionar
– Tiempo de evolución: desarrollo metacrónico de otros tumores
– Cáncer familiar de la enfermedad
– Actuitud agresiva y radical de la tiroides, vaciamiento del compartimiento centra mas linfadenectomia homolateral o incluso contralateral ( esta ultima en un segundo tiempo), por tanto actitud agresiva. A veces incluimos ganglio mediastínicos porque es un tumor que responde mal a la quimioterapia y a la radioterapia.
Debemos de hacer un seguimiento:
– CEA y calcitonina, estos aumentan con recidivas locales, metástasis
– Estudio suprarrenal y paratiroideo ( PTH)
La angina de Ludwig es una infección grave y de rápido progreso que se extiende por el piso de boca y que afecta simultáneamente los espacios submaxilar, sublingual y submentoniano. La infección generalmente comienza con un cuadro agudo de celulitis difusa, manifestándose bilateralmente como una tumefacción de consistencia dura. Su principal origen suele relacionarse a infecciones odontogénicas, sobre todo infección periapical de la segunda y tercera molar inferior (70–80 %), cuyas raíces se extienden debajo de la cresta del músculo milohiodeo de donde se expande la infección hacia otros espacios.
También se han descrito como causas fractura mandibular, sialoadenitis submandibular, infecciones faríngeas o amigdalinas, infecciones a cuerpo extraño o infecciones secundarias a un carcinoma de células escamosas; también se ha relacionado a la  presencia de algunas enfermedades como anemia aplásica, neutropenia, inmunodeficiencias, diabetes o a condiciones como alcoholismo, desnutrición y trasplante de órganos. Los agentes causantes incluyen bacterias aerobias y anaerobias, incluyendo estreptococos y estafilococos, los cuales producen una importante necrosis
presencia de algunas enfermedades como anemia aplásica, neutropenia, inmunodeficiencias, diabetes o a condiciones como alcoholismo, desnutrición y trasplante de órganos. Los agentes causantes incluyen bacterias aerobias y anaerobias, incluyendo estreptococos y estafilococos, los cuales producen una importante necrosis
muscular.
El examen clínico es de extrema importancia, debido a que en la gran mayoría de casos el diagnóstico es alcanzado basándose en los hallazgos clínicos, para ello debemos
seguir los criterios diagnósticos reportados en la literatura como son: celulitis submandibular con proceso gangrenoso y exudado serosanguinolento, que compromete los espacios submandibular, submentoniano y sublingual bilateralmente, pudiendo presentar trismus mandibular, limitación de la apertura bucal, elevación de la lengua, disfagia y odinofagia. Además las personas afectadas muestran una respuesta inflamatoria sistémica que se manifiesta con fiebre, taquicardia y taquipnea; así como protrusión de la lengua con elevación del piso de boca e induración blanda a la palpación y dolor cervical anterior. Se sospecha compromiso del espacio submandibular y la vía aérea, cuando el paciente toma la posición de olfateo para maximizar la entrada
de aire a los pulmones, tiene disfonía, estridor, taquipnea, utiliza los músculos accesorios y maneja mal las secreciones.
Como medio auxiliar de diagnóstico y para guiar un drenaje quirúrgico, se deben solicitar exámenes imagenológicos para valorar las estructuras comprometidas. Se puede solicitar radiografías anteroposteriores de perfil de tórax para buscar evidencias de mayores alteraciones. Esta radiografía puede mostrar el ensanchamiento del espacio mediastínico y la existencia de aire a este nivel. Cuando la infección alcanza el mediastino el paciente presenta dolor retroesternal, disnea severa y tos no productiva. También puede presentar edema y crepitación en el tórax superior. En dichos casos el estado general estará claramente alterado por fiebre elevada, escalofríos y postración extrema.
La radiografía lateral de cuello es útil para observar un desplazamiento de la pared posterior de la faringe, junto con la presencia de gas libre. Una tomografía computarizada es importante porque puede facilitar hallazgos como celulitis en los tejidos blandos de los espacios submandibular y sublingual, colecciones fluidas y formación de abscesos; y además facilita la identificación del compromiso de los espacios laterofaríngeos, retrofaríngeos y mediastino, y la visualización de deformidades o dislocamiento de las vías aéreas. También se pueden solicitar las ultrasonografías
de los tejidos blandos, por ser una modalidad de examen de imagen ideal para diferenciar infecciones superficiales de infecciones profundas más severas.
Complicaciones: La angina de Ludwig es potencialmente letal, debido a que puede extenderse rápidamente al espacio sublingual y submandibular. Por extensión posterior, puede diseminarse al espacio parafaríngeo y por vía anterior al mediastino. El edema de la glotis puede obstruir las vías aéreas superiores y puede resultar imposible el habla, presentándose disfagia y disnea intensa. A la gravedad derivada del cuadro infeccioso que puede traer consigo una sepsis o un shock séptico, se añade el peligro inminente de asfixia además de otras complicaciones como fascitis necrotizante y mediastinitis. El cuadro de mediastinitis descendente necrotizante es una rara pero letal infección de la fascia cervical con compromiso mediastínico, donde la infección progresa a través de los planos faciales cervicales con celulitis, necrosis y formación de abscesos.
Tratamiento Farmacológico y No Farmacológico: El objetivo principal del tratamiento
de la angina de Ludwig es dar soporte a la vía aérea y erradicar el proceso infeccioso mediante la utilización de fármacos y medidas quirúrgicas. Podemos considerar tres aspectos básicos: soporte de la vía aérea, tratamiento farmacológico y procedimientos
quirúrgicos.
Soporte de la vía aérea: El compromiso de la vía aérea se puede presentar hasta en un tercio de los pacientes, y el manejo se debe realizar en aquellos que demuestren edema submandibular con elevación y protrusión de la lengua, estridor, dificultad en el manejo de secreciones, ansiedad y cianosis. Para mantener la vía aérea se puede aplicar un fármaco como la dexametasona 4mg c/6 horas durante 2 días, o cuando no es suficiente utilizar un método más invasivo como la intubación nasotraqueal con visión de fibra óptica. El edema del piso de boca y el trismus generalmente producen dificultades para la intubación, en esos casos se pueden realizar una traqueotomía o cricotiroidectomía. Tratamiento Farmacológico: Las bacterias anaerobias más aisladas son Prevotella, Porphyromonas, Fusobacterium y Peptostreptococos spp; y las bacterias aerobias
son Estreptococos del grupo A, Estreptococos viridans, Staphylococcus aureus y Haemophilus influenzae. Para el tratamiento antibiótico de la angina de Ludwig la mayoría de autores recomiendan: Adultos: Penicilina G 4 a 30 millones de UI al día por vía IV divididas cada 4 o 6 horas o por infusión intravenosa continua; asociada a Metronidazol 1 g de carga seguido de 500mg cada 6 horas por vía IV, aunque otros autores recomiendan 1 g cada 12 horas. Penicilina G cristalina asociada a Clindamicina;
Penicilina – Clindamicina – Amikacina; y Ceftazidima asociada a Clindamicina. Ampicilina con Sulbactam por via IV. Niños: Clindamicina (30 a 40 mg/kg/dia) IV o IM. Ceftazidima (150 mg/kg/dosis) IV. Tratamiento quirúrgico: Está indicado en las siguientes circunstancias: 1) Diagnóstico de celulitis en dos o más de los espacios faciales de cabeza y cuello. 2) Signos clínicos significativos de infección. 3) Infecciones en espacios que pueden comprometer la vía aérea o sean susceptibles de complicaciones. La descompresión quirúrgica está indicada en abscesos de gran tamaño o si después de 24 a 48 horas de antibióticos parenterales no se observa mejoría. En abscesos pequeños la aspiración con aguja es una alternativa al drenaje quirúrgico con antibióticos.

Piso

Piso
contenido:

Contenido
Contenido
Contenido
Triángulo Carotídeo
Contenido
En esta región la carótida interna y externa son fácilmente accesibles (vía clásica de ligaduras de carótida externa).
Contenido:
El triángulo denominado de Guyon es idéntico al de Farabeuf, pero su límite anterosuperior está constituido por el vientre posterior del músculo digástrico, delimitado por:
Contenido
Contenido:
Contenido
La Glándula Tiroides, es un órgano impar, medio simétrico, situado en la cara anterior del cuello, en la unión de su tercio inferior con los dos tercios superiores, se apoya en la parte anterior del conducto laringotraqueal. La tiroides tiene un color gris rosada, consistencia intermedia, mide 7 cm de ancho por 3 de alto y 18 mm de grueso, variando según los individuos, edad y el sexo. Su peso en el adulto, es de 25 a 30 gramos.
Es mantenido en su posición por la cápsula del tiroides que es una extensión de la aponeurosis cervical, posee tres ligamentos; uno medio, que se extiende de la laringe a la parte media del tiroides, y otros laterales, que van de los lóbulos laterales de la tráquea y al cartílago cricoides, también es sostenida por los vasos tiroideos conjuntamente con sus vainas conjuntivas, que de la capsula tiroidea van a la vaina de los vasos del cuello.
Su forma es semejante a un H, cuya concavidad, dirigida hacia atrás, abraza estrechamente los conductos digestivos y respiratorios.
Podemos distinguir una parte media y estrecha el istmo y dos lóbulos laterales más voluminosos.
ISTMO: Tiene 1 cm de alto por 5mm de grueso, sus extremidades laterales se continúan con los lóbulos. Su cara anterior se relaciona con los músculos infrahioideos, la aponeurosis y la piel. Su cara posterior, cóncava, abraza el cricoides y los primeros anillos de la tráquea. Su borde inferior, cóncavo hacia abajo corresponde al segundo anillo traqueal. Su borde superior, cóncavo hacia arriba corresponde al primer anillo de la tráquea. De este borde nace una prolongación en forma de cono, la pirámide de Lalouette, la cual se dirige hacia arriba, costeando uno de los lados del plano medio (mayormente el izquierdo) y se extiende hasta el borde superior del cartílago tiroides; es muy variable en sus dimensiones y en su forma bifurcada en V o en Y invertida; falta en una cuarta parte de los casos; representa morfológicamente la parte inferior del conducto tirogloso, que, en el embrión, une la base de la lengua al vestigio tiroideo medio.
LÓBULOS LATERALES: Cada uno de ellos toma la forma de una pirámide triangular de base inferior, y presenta, por consiguiente, base, vértice, tres caras y tres bordes.
CONSTITUCIÓN ANATÓMICA
La tiroides se compone:
VASOS Y NERVIOS
Las arterias proceden:
1.º de las dos arterias tiroideas superiores, ramas de la carótida externa, cada una de ellas proporcionan tres ramas al cuerpo tiroides: interna, externa y posterior.
2.º de las dos arterias tiroideas inferiores, ramas de la subclavia, cada una de ellas proporciona tres ramas tiroideas: inferior, posterior y profunda.
3.º a veces de una tiroidea media o tiroidea de Neubauer, que nace de la aorta o del tronco braquiocefálico. Las ramificaciones de esas diferentes arterias caminan primero, irregularmente flexuosas, hacia la superficie exterior de la glándula, y después penetran en su espesor, dividiéndose sucesivamente en ramos cada vez más delgados.
Las venas:
Forman alrededor de la glándula un rico plexo: el plexo tiroideo. Las venas que parten de éste se dividen en tres grupos:
1.º venas tiroideas superiores, que corresponden a las arterias del mismo nombre y van a abrirse en la yugular interna, ya sea directamente, ya desaguando previamente en un tronco que les es común con la facial y la lingual: el tronco tirolinguofacial;
2.º venas tiroideas inferiores, que nacen del borde inferior de la tiroides y van a las yugulares internas y al tronco braquiocefálico izquierdo;
3.º venas tiroideas medias, situadas entre las superiores y las inferiores, las cuales van a desaguar en la yugular interna. Es de notar que todas las venas tiroideas son avalvulares.
Los linfáticos:
Forman alrededor de la glándula un plexo peritiroideo. Los troncos que parten de él se dividen en:
1.º linfáticos decendentes, que van a terminar en ganglios situados delante de la tráquea y encima del timo;
2.º linfáticos ascendentes, que terminan en la parte (los medios) en uno o dos ganglios prelaríngeos, y en parte (los laterales) en los ganglios laterales del cuello.
Los nervios:
Proceden:
1.º del simpático cervical

(ganglio cervical medio y segundo nervio cardiaco)
2.º de los dos nervios laríngeos superior recurrente.
Las hormonas tiroideas son determinantes para el desarrollo tanto mental como somático del niño y para la actividad metabólica del adulto. Existen dos tipos de hormonas tiroideas activas biológicamente: la tiroxina (T4), que corresponde al 93% de hormona secretada por la glándula tiroides, y la 3,5,3´-triyodotironina (T3). Ambas están compuestas por dos anillos bencénicos unidos por un puente de oxígeno, uno de los cuales tiene una cadena de alanina y otro un grupo fenilo. La diferencia entre ambas hormonas es que mientras T4 tiene 2 átomos de yodo en el anillo del grupo fenilo, la T3 tiene sólo uno. Existe también otra forma denominada rT3 (3,3´,5´ triyodotironina inversa) que no posee actividad biológica.
Metabolismo del yodo:
Para formar una cantidad normal de tiroxina se precisan al año unos 50 mg de yodo (ingerido en forma de yoduros), o sea, unos 150mg/día en adultos. La cantidad necesaria es mayor en embarazadas, unos 220 mg/día, y en niños varía con la edad. Si las cantidades ingeridas son crónicamente inferiores aparece bocio (aumento del tamaño de la glándula). Lo mismo ocurre al ingerir sustancias que interfieren en la absorción  gastrointestinal del yodo o bien en su utilización por la glándula denominadas bociógenos. Para evitar el déficit de yodo se ha añadido yoduro sódico a la sal común. Los yoduros ingeridos por vía oral se absorben desde el tubo digestivo hasta la sangre. La mayoría se excreta vía renal, pero, en condiciones normales, 1/5 parte es retirada por las células tiroideas para la síntesis de hormonas tiroideas. Para medir el déficit de yodo se puede medir la excreción urinaria del mismo, así, a menor excreción, mayor déficit. Por otra parte, y en sentido inverso, también las hormonas tiroideas son metabolizadas hasta yoduros en diversos tejidos diana de las mismas. Este yoduro pasa a sangre y de nuevo es captado por la glándula tiroides o excretado por orina. Existe una pequeña cantidad de yodo (unos 10-20mg) que se pierde por las heces. Cuando la ingesta de yodo es inferior a los requerimientos aumenta la proporción que es captada y utilizada en la tiroides frente a la que se elimina por la orina. Cuando la ingesta es superior a los requerimientos se elimina una proporción mayor por la orina.
gastrointestinal del yodo o bien en su utilización por la glándula denominadas bociógenos. Para evitar el déficit de yodo se ha añadido yoduro sódico a la sal común. Los yoduros ingeridos por vía oral se absorben desde el tubo digestivo hasta la sangre. La mayoría se excreta vía renal, pero, en condiciones normales, 1/5 parte es retirada por las células tiroideas para la síntesis de hormonas tiroideas. Para medir el déficit de yodo se puede medir la excreción urinaria del mismo, así, a menor excreción, mayor déficit. Por otra parte, y en sentido inverso, también las hormonas tiroideas son metabolizadas hasta yoduros en diversos tejidos diana de las mismas. Este yoduro pasa a sangre y de nuevo es captado por la glándula tiroides o excretado por orina. Existe una pequeña cantidad de yodo (unos 10-20mg) que se pierde por las heces. Cuando la ingesta de yodo es inferior a los requerimientos aumenta la proporción que es captada y utilizada en la tiroides frente a la que se elimina por la orina. Cuando la ingesta es superior a los requerimientos se elimina una proporción mayor por la orina.
El simportador na +/I:
El primer paso en la formación de hormonas tiroideas consiste en el trasporte de los yoduros desde la sangre hasta las células y folículos tiroideos. El transporte de yodo al interior de la célula se produce en contra de gradiente electroquímico y tiene lugar gracias a una proteína transmembrana localizada en la membrana basolateral de las células foliculares tiroideas denominada simportador Na+/I- (NIS). Se produce por un proceso de transporte activo secundario, la energía es proporcionada por el transporte de Na+ hacia el exterior de la célula mediante la ATP-asa de Na+ y K+. Este mecanismo es capaz de producir concentraciones intracelulares de I- que son de 20-40 veces mayores que la concentración plasmática. El principal regulador de la actividad del NIS es la hormona estimuladora del tiroides (TSH). Otros iones tales como el perclorato y pernectato son también transportados al interior de la glándula tiroides por el mismo mecanismo actuando, así como inhibidores competitivos del transporte de yodo.
La tiroglobulina (TG) es una glucoproteína de gran peso molecular (660 kDa) compuesta por 2 subunidades idénticas unidas por enlaces no covalentes. Se encuentra  mayoritariamente en el lumen de los folículos tiroideos. El retículo endoplasmático y el aparato de Golgi son los encargados de sintetizar y glicosilar la TG y secretarla hacia los folículos. Las moléculas de TG glicosilada se empaquetan en vesículas exocitócicas, saliendo así del aparato de Golgi al citoplasma celular. Estas vesículas se funden en la membrana apical que bordea el lumen folicular, liberando su contenido al mismo. Tanto la síntesis de TG como su exocitosis al lumen están bajo el control de la TSH.
mayoritariamente en el lumen de los folículos tiroideos. El retículo endoplasmático y el aparato de Golgi son los encargados de sintetizar y glicosilar la TG y secretarla hacia los folículos. Las moléculas de TG glicosilada se empaquetan en vesículas exocitócicas, saliendo así del aparato de Golgi al citoplasma celular. Estas vesículas se funden en la membrana apical que bordea el lumen folicular, liberando su contenido al mismo. Tanto la síntesis de TG como su exocitosis al lumen están bajo el control de la TSH.
Cada molécula de TG contiene unos 110-120 residuos del aminoácido tirosina, que es el sustrato principal que se combina con el yodo en un proceso denominado organificación de la tiroglobulina para dar lugar a las hormonas tiroideas. Así pues, las hormonas tiroideas se forman dentro de la molécula de TG. Para que los iones yoduro se puedan unir a la tirosina han de pasar a una forma oxidada del yodo. Este proceso de oxidación tiene lugar gracias a la enzima peroxidasa y su peróxido de hidrógeno acompañante necesario para la reacción. Esta enzima se encuentra en la membrana apical de la célula tiroidea, proporcionando así el yodo oxidado justo en el lugar donde la molécula de TG abandona el aparato de Golgi. Esta peroxidasa cataliza la yodación de aproximadamente el 10% de los residuos de tirosina de la TG. En el proceso de síntesis hormonal, el primer producto es la monoyodotirosina (MIT). Ésta se une con un nuevo yodo en posición 5 para formar diyodotirosina (DIT). Las moléculas de DIT y MIT se unen entre sí mediante un proceso denominado reacción de acoplamiento.
El principal producto hormonal de la reacción de acoplamiento es la molécula de tiroxina (T4), que resulta de la unión de 2 moléculas de DIT, y que aún forma parte de la molécula de tiroglobulina. En otras ocasiones DIT se une a MIT para formar triyodotironina (T3). En condiciones normales una molécula de TG contiene unas 6 moléculas de MIT, 4 de DIT, 2 de T4 y 0.2 de T3. Sólo existen trazas de rT3 y otros componentes. Si la concentración de yoduro es más baja, no se alcanza el grado de yodación de la TG necesario para la formación de T4, ya que se forman menos residuos de DIT que de MIT. En este caso se favorece la formación de T3, con lo que se forma una molécula más activa biológicamente. Este proceso se conoce como síntesis preferente de T3, y facilita la adaptación a situaciones de ingesta de yodo insuficiente.
Secreción de las hormonas tiroideas
La TG permite almacenar en los folículos una cantidad de hormona tiroidea suficiente para cubrir las necesidades normales del organismo durante 2 o 3 meses. Para poder liberar T3 y T4, la TG ha de ser reabsorbida por la célula tiroidea. La TG entra al citoplasma mediante un proceso de macropinocitosis, pero sobre todo por micropinocitosis. La superficie apical de las células tiroideas emite extensiones en forma de seudópodos que rodean pequeñas porciones de coloide, constituyendo vesículas de pinocitosis. Éstas se unen a lisosomas del citoplasma celular dando lugar a fagolisosomas. Los lisosomas contienen unas proteinasas, las catepsinas B, L y D, que permiten la proteólisis de la TG. La digestión de la TG deja T3 y T4 intactas, que pasan al torrente circulatorio, mientras que DIT y MIT son retenidas y desyodadas para ser recicladas dentro de la célula. La desyodación de DIT y MIT tiene lugar gracias a la acción de una enzima denominada yodotirosina desyodasa o deshalogenasa. La enzima que desyoda las yodotirosinas DIT y MIT es diferente de las enzimas que desyodan las yodotironinas T4 y T3. La mayoría de este yodo liberado es reutilizado por la glándula para formar nuevas hormonas tiroideas. Respecto a la tiroxina, no toda la T4 liberada por hidrólisis sale a la sangre. Parte de T4 se convierte en T3 gracias a la acción de una yodotironina desyodasa que tiene la particularidad de ser estimulada por la TSH. En condiciones normales, alrededor del 93% de la hormona tiroidea liberada por el tiroides corresponde a T4 y sólo el 7% es T3.
Más del 99,95% de T4 y 99,5% de T3 están unidas a proteínas en sangre como son: la globulina fijadora de tiroxina (TBG), transtirretina (TTR, anteriormente llamada prealbúmina fijadora de tiroxina o TBPA), albúmina y lipoproteínas. La vida media de la TTR es de 2 días, la de la TBG 5 días y la de la albúmina.
 lipoproteínas. Aproximadamente el 0.02 % está libre en suero.
lipoproteínas. Aproximadamente el 0.02 % está libre en suero.La concentración de T4 y T3 libres es lo que determina la actividad biológica de estas hormonas y está controlada de manera muy precisa. Por ejemplo, cuando existe un aumento en la concentración de proteínas de unión en el plasma, la concentración de hormonas libres disminuye. Este descenso estimula la secreción de TSH hipofisaria que, a su vez incrementa la producción de hormonas libres.
Tiroxina (T4):
La producción de hormonas tiroideas se produce íntegramente en la glándula tiroidea y es de 100-130 nmoles/día 14. La reserva extratiroidea de T4 es de 1000-1300 nmoles, la mayoría extracelular. La T4 se degrada un 10% al día. El 80% es desyodada, un 40% para formar T3 y el otro 40% para formar rT3. El 20% restante o bien se conjuga con glucurón y sulfato, o sufre desaminación o descarboxilación en la cadena de alanina formándose sus derivados acéticos y propiónicos respectivamente. La formación de glucuronoconjugados y sulfatoconjugados de T3 y T4 tiene lugar principalmente en el hígado y en el riñón. En el caso del hígado son excretados por la bilis al intestino, en donde son hidrolizados, volviendo a ser absorbidos como T4 y T3, o eliminados como tales conjugados por las heces (circulación enterohepática). Esta vía es relativamente poco importante en el ser humano. La vía más importante de metabolización de T4 y T3 es la desyodación en cascada de la molécula. La pérdida de un átomo de yodo en la posición 5´ de T4 da lugar a la formación de T3, que es más activa biológicamente. Si la pérdida de yodo es en la posición 5 se forma rT3 (inactivación de la T4).
Triyodotironina (T3):
Más del 80% de T3 se produce por desyodación extratiroidea de T4 y el resto se forma directamente por la tiroides14. La producción total de T3 es 45-60 nmoles/día. La reserva extratiroidea de T3 es de 75 nmoles, la mayoría intracelular. T3 se degrada mayoritariamente por desyodación a una velocidad mucho mayor que T4, un 75% al día 3.
Triyodotironina reversa (rT3):
La producción de rT3 es 45-60 nmoles/día, por desyodación extratiroidea de T4 14. La rT3 se degrada por desyodación a una velocidad más rápidamente que T3.
Desyodación en cascada
Tal y como hemos comentado, la desyodación en cascada supone la vía metabólica más importante de las hormonas tiroideas. La desyodación de T3 y T4 se produce en el hígado, riñones y muchos otros tejidos. Existen diferencias entre la proporción T3/T4 en distintos tejidos. Una proporción muy alta de T3/T4 hay en hipófisis y córtex cerebral. Existen 3 tipos de desyodasas que mantienen el índice T3/T4 en los tejidos: DI, DII y DIII. Todas contiene el raro aminoácido selenocisteína, y el selenio es esencial para su actividad enzimática.
DI: se encuentra en hígado, riñones, tiroides e hipófisis. Desyoda en el siguiente orden: rT3>T4>T3. Es inhibida por propiltiouracilo (PTU sensible).
DII: está en cerebro, hipófisis, músculo, piel, placenta y grasa parda; también contribuye a la formación de T3. Desyoda T4>rT3. No inhibida por PTU.
DIII: presente principalmente en cerebro, piel y placenta. Actúa sobre la posición 5 de T3 y T4, y es probable que sea la fuente principal de rT3 de sangre y tejidos.
El 80% de T3, que es la hormona con mayor actividad biológica, se produce en tejidos extratiroideos gracias a las desyodinasas DI y DII, que están, respectivamente, en la membrana plasmática y en los enzimas microsomales.
En sujetos normales, el 65% de T3 producida de forma extratiroidea se debe a DII y el resto a la DI. La proporción en la que contribuye DII es mayor en el hipotiroidismo y menor en el hipertiroidismo.
La existencia de una cantidad adecuada de hormona tiroidea en el organismo se regula a través del hipotálamo y de la adenohipófisis que controlan la secreción tiroidea. Estos mecanismos se explican a continuación:
La TSH, o tirotropina, en una hormona adenohipofisaria que aumenta la secreción de T3 y T4 por la glándula tiroidea. La TSH:
La secreción de TSH por la hipófisis está controlada por una hormona hipotalámica, la hormona liberadora de tirotropina (TRH), transportada hasta la adenohipófisis por la circulación portal hipotálamo-hipofisaria.
Uno de los estímulos que más aumentan la secreción de TRH y, por consiguiente, la de TSH, es la exposición al frío, en un control fisiológico de la temperatura por los centros hipotalámicos. Sustancias como la somatostatina o la dopamina también aumentan estimulan la cascada desde hipotálamo. Los estados de ansiedad disminuyen la secreción de TSH.
El aumento de hormona tiroidea en sangre reduce la secreción de TSH. Cuando la secreción de hormona tiroidea aumenta hasta 1.75 veces del valor normal, la secreción de TSH disminuye prácticamente hasta desaparecer, por acción directo sobre la propia adenohipófisis.
Las hormonas tiroideas ejercen su acción tras su introducción en el interior de la célula. En los últimos años han sido clonados e identificados dos tipos distintos de receptores nucleares de las hormonas tiroideas (TRa y TRb) codificados por genes localizados, respectivamente, en los cromosomas 17 y 3. La unión de la T3 con estos receptores nucleares origina el complejo T3TR, el cual, a su vez, funciona uniéndose a secuencias específicas de DNA o elementos de respuesta que se encuentran en las zonas reguladoras de genes que responden a las hormonas tiroideas. La T3 controla la expresión de numerosos genes que a su vez regulan la síntesis de diversas proteínas. Además de este mecanismo central, las hormonas tiroideas poseen un efecto calorígeno y también un efecto primario sobre la membrana citoplasmática, regulando el flujo transcelular de sustratos y cationes.
A través de los citados mecanismos de acción, de gran complejidad, las hormonas tiroideas activan el metabolismo energético, incrementando el consumo calórico, regulan el crecimiento y maduración de los tejidos y el recambio de prácticamente todos los sustratos, vitaminas y hormonas.
Se trata de una glándula con arquitectura folicular característica. A pequeños aumentos se distinguen estructuras foliculares de distintos tamaños con un contenido eosinófilo denominado coloide. Los distintos folículos están acompañados de un conectivo rico en vasos sanguíneos. Los folículos de gran tamaño aparecen distendidos por su contenido coloide y tapizados por un epitelio simple aplanado, mientras que los folículos de pequeño tamaño presentan un clásico epitelio cúbico bien definido. Alrededor de determinados folículos vemos algunos acúmulos de células claras, correspondientes a las células C o parafoliculares. El estroma interfolicular presenta células que en ocasiones corresponden a una visión de acúmulos de celulas C que revisten el foliculo pero al corte quedan dando una falsa visión de células localizadas entre folículos.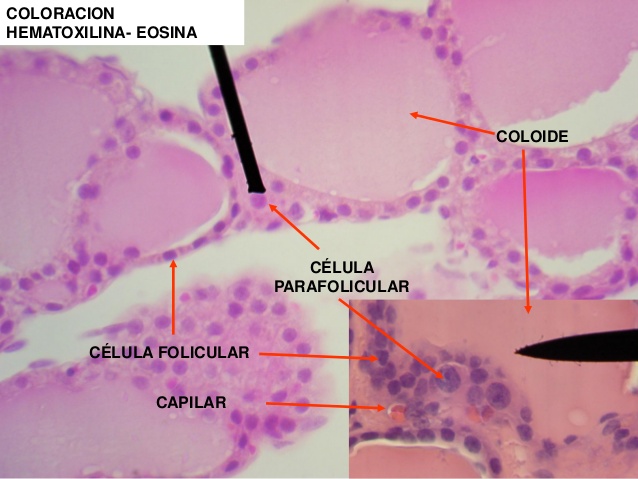
Sistema ABO
El sistema ABO es el primer sistema de grupo sanguíneo humano descubierto (Landsteiner 1900). Los cuatro grupos sanguíneos de este sistema, AB, A, B y O, están determinados por la presencia o no de dos antígenos, denominados A y B en la membrana del eritrocito. Los antígenos del sistema ABO no se hallan circunscriptos a los eritrocitos, sino que se encuentran también en leucocitos, plaquetas y células de tejidos. Asimismo se encuentran sustancias activas de grupo sanguíneos en la mayoría de los líquidos orgánicos. Se dice que las personas cuyos líquidos orgánicos contienen sustancias de grupo sanguíneo son secretoras; aquellas cuyos líquidos orgánicos no contienen sustancias de grupo sanguíneo se denominan no secretoras. Los eritrocitos transportan una rica variedad de antígenos individuales. Algunos de ellos, aunque altamente inmunógenos, son tan frecuentes (públicos) o tan raros (privados) que rara vez están involucrados en reacciones adversas, aunque pueden ser responsables de la inmunoacción sobre un feto o contra células transfundidas. Las reacciones severas por transfusiones habitualmente se deben a incompatibilidad para los antígenos del sistema ABO. Estos antígenos son oligosacáridos y están codificados genéticamente por genes situados en locus separados. Un gen H situado en otro locus, codifica la sustancia precursora sobre la que actúan los productos de los genes A y B. Estos productos son enzimas que actúan como transferasas específicas. El producto del gen H es una enzima que produce la sustancia H. Las transferasas de los genes A y B son enzimas que convierten la sustancia H, en antígenos A y B. En otras palabras, la sustancia H es la base sobre la cual actúan los genes A y B para formar los antígenos A y B. Por lo tanto las células del grupo O están dotadas generosamente de sustancia H, mientras que en las células A y B la mayor parte del sustrato se utiliza, de manera que queda relativamente poca sustancia H. El gen O es un alelo silencioso (no altera la estructura de la sustancia H). De los individuos que no heredan el gen H, se dice que pertenecen al fenotipo Bombay (hh); estos individuos no producen sustancia H y, como consecuencia los genes A y B, si los tienen, no pueden expresarse.
La mayoría de los eritrocitos humanos contiene sustancia H, que se encuentra en asociación con los principales grupos sanguíneos en el siguiente orden decreciente de concentración: O, A2, A2B, B, A1, A1B. El gen H se hereda independientemente de los grupos ABO y del estado secretor. Los genotipos HH y Hh son H – positivos, hh es H – negativo (grupo Oh o grupo Bombay).
Bioquímica
Los antígenos A, B, H del plasma así como los de los hematíes, son glucoproteínas y glucolípidos. Otros autores consideran que las sustancias de los grupos sanguíneos son glucolípidos sobre los eritrocitos, pero aparecen como glucoproteínas en la forma secretoria que se encuentra en los líquidos orgánicos. La especificidad antigénica está determinada por los glúcidos en los extremos no reductores del componente hidrato de carbono.
Los principales determinantes estructurales de la especificidad de los grupos H, A y B son los siguientes:
Los productos primarios del gen del locus de grupo sanguíneo son glucosil-transferasas, que dirigen el agregado de las unidades de glúcidos adecuadas a los sustratos aceptores preformados. Las enzimas son específicas, no sólo para el tipo de glúcido agregado, sino también para el sustrato y el tipo de unión.
Fenotipo Bombay
Los individuos que poseen este raro fenotipo, no heredan el gen H (son hh) y no son capaces de producir la sustancia H; pueden sí heredar los genes A y B pero por su carencia de sustancia H, no pueden producir los antígenos A y B. Los hematíes del fenotipo Bombay son aparentemente como los del fenotipo O, pero la diferencia es la carencia de la sustancia H en los individuos Bombay. Los hijos de estos individuos, pueden expresar cantidades normales de los antígenos A y/o B en sus hematíes, siempre que hereden un gen H del otro progenitor; de este modo, dos individuos cuyo fenotipo es O en apariencia, pueden tener un descendiente con fenotipo A y/o B.
Subgrupos
El fenotipo A puede dividirse en dos subgrupos. Aproximadamente el 80% de los individuos del grupo A tienen el fenotipo A1 y el 20% restante el fenotipo A2. Entre estos dos subgrupos existen diferencias cualitativas y cuantitativas. Los individuos A1 producen antígeno A a partir de todas las cadenas H de tipo II (H1, H2, H3, H4). Los individuos A2 producen antígeno A solamente a partir de los precursores H1 y H2. Por lo tanto, los individuos A1 presentan más cantidad de antígeno A por eritrocito que los individuos A2. Aproximadamente el 3% de los individuos A2 y el 25% de los individuos A2B producen un anticuerpo denominado anti A1. Este anticuerpo reacciona con los hematíes A1, pero no reacciona con los hematíes A2. Es posible que el anti – A1 reaccione con el antígeno A que se forma a partir de las cadenas H3 y H4. Los hematíes pertenecientes al subgrupo A3, presentan un modelo característico de aglutinación cuando reaccionan con el suero anti – A: algunos de los hematíes son aglutinados mientras que otros no lo son, es decir, ofrecen una imagen de doble población. El fenotipo A3 presenta una frecuencia de 1:1000.
Subgrupos más raros de A son: Ax, Am, Aend, Ael y AFinn. Recientemente, aparece un subgrupo denominado Aint (A intermedio) que comparte ciertos caracteres con A1 y A2. El reconocimiento de variantes débiles del grupo A reviste importancia cuando se presentan reacciones transfusionales hemolíticas y en la práctica forense. Es importante la identificación de los donantes de sangre que pertenecen a estos subgrupos A con el objeto de evitar que sean erróneamente etiquetados como donantes del grupo sanguíneo O. Si se transfundiera sangre a uno de los subgrupos de A indicados a un receptor del grupo O, podría tener lugar una reacción transfusional. Los subgrupos de B son menos comunes. Entre ellos: B3, Bx, Bm y Bel, pero de muy baja frecuencia.
Anticuerpos del sistema ABO
Los anticuerpos anti A y anti B, son producidos por individuos que carecen de los antígenos A y B respectivamente, de acuerdo con la regla que un antígeno y su anticuerpo correspondiente nunca se encuentran juntos en la sangre de la misma persona. Dichos anticuerpos son predominantemente del tipo IgM y también (menos frecuentes y de carácter inmunogénico), del tipo IgG. Este tipo de anticuerpos, puede ser producido por individuos del grupo O. Los anticuerpos del sistema ABH, son denominados «naturales», ya que aparecen en las primeras etapas de la vida extrauterina, por exposición a antígenos ubicuos presentes en superficies bacterianas y ciertos tipos de alimentos, que tienen una composición similar a los antígenos presentes en la membrana de los eritrocitos y producen inmunización. Lógicamente, el reconocimiento primitivo de lo propio a cargo del sistema inmunológico, hace que los anticuerpos que se produzcan no sean de los antígenos correspondientes al mismo individuo. El anti A – B perteneciente al grupo O, no es una simple mezcla de anti – A y de anti – B sino que es un tercer anticuerpo que presenta reacción cruzada con un antígeno presente en los hematíes A y B; este antígeno es denominado complejo A, B o antígeno C. Los anticuerpos del sistema ABH, pueden reaccionar a la temperatura corporal y activar al complemento, causando una rápida destrucción intravascular de los hematíes. El anti – H puede presentarse como un autoanticuerpo natural en el suero de individuos A, A – B o B, o bien como un aloanticuerpo en el plasma de los individuos del fenotipo Bombay. En este caso, su rango térmico es elevado lo cual, junto con su capacidad para fijar el complemento, hace que el anticuerpo anti – H sea clínicamente significativo; por lo tanto, los individuos Bombay, sólo pueden ser transfundidos con sangre de otros individuos pertenecientes a dicho fenotipo.
SISTEMA RHESUS
Representa otro sistema de antígenos de eritrocitos, que no está químicamente caracterizado. En 1940 Landsteiner y Wiener efectuaron comunicaciones en el sentido que si inyectaban eritrocitos de mono Rhesus a conejos o cobayos, estos animales producían un anticuerpo que, después de su absorción, aglutinaban los eritrocitos de un 85% aproximadamente, de personas norteamericanas de raza blanca. Denominaron a este anticuerpo anti – Rh (Rhesus) y el antígeno que se detectaba recibió el nombre de antígeno Rh. Poco antes de esto Levine y Stetson habían encontrado un anticuerpo en el suero de una mujer del grupo O, que antes no había sido transfundida, que presentó una reacción después de recibir una transfusión de sangre del Grupo O de su marido. Más tarde la paciente dio a luz un feto macerado, y estos autores sugirieron que había producido un anticuerpo para un antígeno eritrocítico fetal heredado del marido. Al parecer los anticuerpos humanos y animales eran idénticos, y por lo tanto se aceptó el nombre de anti – Rh para el anticuerpo humano. Más tarde se vio que los dos anticuerpos no eran iguales y por tal motivo continuó denominándose anti – Rh al anticuerpo humano y se le dio el nombre de anti – LW al anticuerpo animal en honor a Landsteiner y Wiener, sus descubridores.
Los antígenos del sistema Rh son algunas veces responsables de reacciones por transfusión menos severas que el ABO. Son proteínas y rara vez se encuentran en el medio, de modo que los anticuerpos preformados son raros. Los genes que codifican los antígenos del sistema Rh están localizados en el brazo corto del cromosoma 1.
El sistema Rhesus está constituido por unos 40 antígenos distintos, 5 de los cuales revisten importancia especial.
Existen dos nomenclaturas para designar los distintos antígenos del sistema Rh:
Variantes
Existe una variante débil del antígeno D (en menor importancia C o E), denominado Du (Cu o Eu ). Se encuentra en algunos individuos, aunque raros, en cuyas células faltan algunos (- D -) o todos (—) los antígenos Rh. En estos últimos casos, el estado se denomina Rh – nulo. Dichas células tienen un tiempo de sobrevida corta y quizás poseen un defecto estructural básico en su membrana. Estas células son útiles para el estudio de la anemia hemolítica autoinmune.
Antígeno D
Es una variante débil del antígeno D, poco frecuente entre los individuos caucasoides, pero común entre los individuos de raza negra (22%).
Los hematíes D generalmente dan reacciones débiles o negativas con el anticuerpo anti – D, siendo detectados gracias a la prueba indirecta de la antiglobulina (Coombs).
Los hematíes D pueden ser clasificados de acuerdo a tres categorías:
Hematíes con delección Rh (-D-) y Rh nulo (—)
De los hematíes que no poseen los antígenos dependientes de los locus C y E, se dice que presentan delección (-D-). El número de lugares antigénicos D está aumentado en estos hematíes, y por lo tanto el anti – D de tipo IgG puede aglutinarlo. De los individuos que no expresan ninguno de los antígenos del sistema Rhesus en sus hematíes, se dice que tienen Rh nulo (—). Los eritrocitos de estos individuos tienen alterado el transporte de Na+ y K+. Esto da lugar a una anemia
Anticuerpos del sistema Rhesus
Son extraordinariamente importantes en medicina clínica. Los anticuerpos del sistema Rhesus se producen en forma de anticuerpos completos (IgM o incluso IgA), o lo que es más común, como anticuerpos incompletos (IgG) siendo estimulada su producción por transfusión o por embarazo. No activan al complemento debido a que la situación de los antígenos Rhesus en la membrana de los hematíes no permite la formación de dobletes de IgG necesarios para la activación del mismo. Los anticuerpos del sistema Rh pueden causar reacciones transfusionales y enfermedad hemolítica del recién nacido.
Los anticuerpos completos son anticuerpos salinos porque aglomeran los hematíes suspendidos en una solución de NaCl o en un medio con alta concentración proteica y también reciben el nombre de anticuerpos bivalentes, aglutinantes o inmunes tempranos, porque son los primeros en aparecer; son detenidos por la placenta intacta y el papel que desempeñan en la eritroblastosis fetal es secundario.
Los anticuerpos incompletos son también llamados de bloqueo, monovalentes, de albúmina, conglutinantes e hiperinmunes; producen aglomeración solamente cuando en lugar de una solución salina, se emplea un medio adecuado de proteína. Son de aparición tardía, pasan fácilmente a través de la placenta intacta y desempeñan un papel muy importante en la eritroblastosis fetal.
Los anticuerpos del tipo IgG se combinan con los sitios del antígeno en la superficie del eritrocito pero son demasiado pequeños como para causar aglutinación a menos que el estado normal de repulsión entre los eritrocitos (ver potencial zeta), se encuentre reducido por descenso de la carga negativa. Esto puede lograrse si se trata a las células con ciertas enzimas proteolíticas (por ejemplo tripsina, papaína, ficina, etc.) o si se suspenden las células en albúmina bovina al 20 o 30%. Esta última actúa elevando la constante dieléctrica del medio eritrocítico. El medio estándar para demostrar la presencia de los anticuerpos incompletos es la prueba de Coombs.
Transfusión de sangre
La única indicación para la transfusión de sangre completa es la hemorragia, ya sea posterior a trauma, durante algún tratamiento quirúrgico o asociado a sangrado gastrointestinal masivo u otro sangrado. La tipificación sistemática de los tipos sanguíneos y la compatibilidad cruzada entre la sangre del donador y la del receptor son practicadas antes de la administración de cualquier producto derivado de la sangre que contenga eritrocitos.
Los eritrocitos empaquetados se administran para restaurar la masa eritrocítica en los enfermos con anemias crónicas sin hipovolemia. Las ventajas de esta administración en lugar de la sangre total, incluyen las siguientes:
Las transfusiones de sangre no deben ser efectuadas como sustituto para una investigación completa de un enfermo con anemia, ni para evitar la corrección de anormalidades específicas, por ejemplo la deficiencia de vitamina B12 o deficiencia de hierro.
Las indicaciones especiales para la transfusión de sangre son:
Autotransfusión
La mayor parte de los riesgos de la transfusión sanguínea como la incompatibilidad, la aloinmunización, la inmunosupresión y la transmisión de enfermedades infecciosas desaparecen con la autotransfusión, a la vez que ayuda a mantener el abastecimiento del banco de sangre. Autodonación y predepósito: Se utiliza en cirugías programables que suelan necesitar transfusión, autodonando el paciente aproximadamente una vez por semana si lo tolera, mientras recibe suplementos de hierro. Se pueden conseguir hasta 6 unidades. Una variante de la misma es la hemodilución normovolémica en la que se extrae el paciente 24-48 horas antes de la intervención, una o dos unidades de sangre sustituyendo su volumen por soluciones coloides o cristaloides; con ello se mejora la microcirculación y se reduce la pérdida de hematíes en un volumen dado de hemorragia intraoperatoria.
Reacciones de las transfusiones de sangre
Son debidas a mecanismos no inmunitarios y debidas a mecanismos inmunitarios. Las primeras incluyen las anormalidades hemodinámicas debidas a la sobrecarga líquida, trastornos hemostáticos por transfusiones masivas, organismos infectantes en la sangre del donador (especialmente el antígeno HBs de la hepatitis y HIV del síndrome de inmunodeficiencia adquirida) y otras complicaciones incluyendo embolia y tromboflebitis.
Las reacciones inmunitarias incluyen las reacciones alérgicas, la incompatibilidad de los eritrocitos, las incompatibilidades asociadas con los leucocitos, las plaquetas y las reacciones de los aloanticuerpos.
Las reacciones más graves con hemólisis intravascular, usualmente son debidas a incompatibilidad ABH (ABO). El enfermo presenta fiebre, malestar, escalofríos y dolor de espalda poco tiempo después que comienza la transfusión, pudiendo entrar en choque. La insuficiencia renal aguda es el riesgo más grande.
La causa más común de hemólisis extravascular es la incompatibilidad del antígeno Rh con los anticuerpos anti Rh0 (D) (anti – Rh). Los síntomas y signos son menos intensos y su comienzo puede ser más tardío. La insuficiencia renal aguda es menos común. Las reacciones retardadas de la transfusión por incompatibilidad Rh pueden no ser evidentes durante 4 – 14 días después de la transfusión.
Las reacciones a los leucocitos y las plaquetas implican anticuerpos para el sistema HLA y otros aloantígenos de la membrana celular. Estas reacciones producen fiebre y escalofríos, pero habitualmente no causan otras complicaciones graves. Debe emplearse sangre especialmente preparada como eritrocitos pobres en leucocitos o congelados y lavados, cuando se transfunda a enfermos que están sensibilizados a estos antígenos.
Los anticuerpos para las inmunoglobulinas séricas del hombre son hallados en ciertos enfermos y los anticuerpos anti – IgA son de importancia en medicina. Los enfermos con deficiencia selectiva de IgA o (rara vez) hipogammaglobulinemia, pueden elaborar anticuerpos anti – IgA. Estos anticuerpos incluyen diversas especificidades que reaccionan con la clase IgA, las subclases IgA y los alotipos IgA de anticuerpos. La exposición posterior de dicho enfermo sensibilizado a la sangre o productos de la misma que contengan IgA, puede producir reacción urticarial o hasta una reacción anafiláctica mortal. .
En función de su distribución corporal, las soluciones intravenosas utilizadas en fluidoterapia pueden ser clasificadas en: 1) Soluciones cristaloides y 2) Soluciones coloidales.
SOLUCIONES CRISTALOIDES.
Las soluciones cristaloides son aquellas soluciones que contienen agua, electrolitos y/o azúcares en diferentes proporciones y que pueden ser hipotónicas, hipertónicas o isotónicas respecto al plasma.
Soluciones cristaloides isoosmóticas.
Dentro de este grupo las que se emplean habitualmente son las soluciones salina fisiológica (ClNa 0.9 %) y de Ringer Lactato que contienen electrolitos en concentración similar al suero sanguíneo y lactato como buffer.
La solución salina al 0.9 % también denominada Suero Fisiológico, es la sustancia cristaloide estándar, es levemente hipertónica respecto al líquido extracelular y tiene un pH ácido. La relación de concentración de sodio (Na+) y de cloro (Cl) que es 1/1 en el suero fisiológico, es favorable para el sodio respecto al cloro (3/2) en el líquido extracelular (Na+ > Cl). Contiene 9 gramos de ClNa o 154 mEq de Cl y 154 mEq de Na+ en 1 litro de H2O, con una osmolaridad de 308 mOsm/L.
Estas soluciones cristaloides no producen una dilución excesiva de factores de coagulación, plaquetas y proteínas, pero en déficits severos se puede producir hipoalbuminemia, con el consecuente descenso de la presión coloidosmótica capilar (pc) y la posibilidad de inducir edema. Este descenso de la pc, con su repercusión en gradiente transcapilar, atribuído a la administración excesiva de soluciones cristaloides, ha sido considerada como favorecedor de la formación de edemas.
La solución de Ringer Lactato contiene 45 mEq/L de cloro menos que el suero fisiológico, causando sólo hipercloremia transitoria y menos posibilidad de causar acidosis. Y por ello, es de preferencia cuando debemos administrar cantidades masivas de soluciones cristaloides. Diríamos que es una solución electrolítica “balanceada”, en la que parte del sodio de la solución salina isotónica es reemplazada por calcio y potasio.
La solución de Ringer Lactato contiene por litro la siguiente proporción iónica: Na+= 130 mEq, Cl = 109 mEq, Lactato= 28 mEq, Ca2+ = 3 mEq y K+ = 4 mEq.Estas proporciones le supone una osmolaridad de 273 mOsm/L, que si se combina con glucosa al 5 % asciende a 525 mEq/L. El efecto de volumen que se consigue es muy similar al de la solución fisiológica normal.
La infusión de Ringer Lactato, contiene 28 mEq de buffer por litro de solución, que es primeramente transformado en piruvato y posteriormente en bicarbonato durante su metabolismo como parte del ciclo de Cori.
Solución Salina Hipertónica.
Las soluciones hipertónicas e hiperosmolares han comenzado a ser más utilizados como agentes expansores de volumen en la reanimación de pacientes en shock hemorrágico.
Entre sus efectos beneficiosos, además del aumento de la tensión arterial, se produce una disminución de las resistencias vasculares sistémicas, aumento del índice cardíaco y del flujo esplénico.
El mecanismo de actuación se debe principal y fundamentalmente, al incremento de la concentración de sodio y aumento de la osmolaridad que se produce al infundir el suero hipertónico en el espacio extracelular (compartimento vascular). Así pues, el primer efecto de las soluciones hipertónicas sería el relleno vascular. Habría un movimiento de agua del espacio intersticial y/o intracelular hacia el compartimento intravascular. Recientemente se ha demostrado que el paso de agua sería fundamentalmente desde los glóbulos rojos y células endoteliales (edematizadas en el shock) hacia el plasma, lo que mejoraría la perfusión tisular por disminución de las resistencias capilares. Una vez infundida la solución hipertónica, el equilibrio hidrosalino entre los distintos compartimentos se produce de una forma progresiva y el efecto osmótico también va desapareciendo de manera gradual.
Otros efectos de la solución hipertónica son la producción de hipernatremia (entre 155-160 mmol/L) y de hiperosmolaridad (310-325 mOsm/L). Esto puede ser de suma importancia en ancianos y en pacientes con capacidades cardíacas y/o pulmonares limitadas. Por ello es importante el determinar el volumen máximo de cloruro sódico que se puede administrar, ya que parece deberse a la carga sódica el efecto sobre dichos órganos. También se ha demostrado que la perfusión de suero hipertónico eleva menos la PIC (Presión Intracraneal).
Soluciones de comportamiento similar al agua.
Se clasifican en glucídicas isotónicas o glucosalinas isotónicas.
Es una solución isotónica (entre 275-300 mOsmol/L) de glucosa, cuya dos indicaciones principales son la rehidratación en las deshidrataciones hipertónicas (por sudación o por falta de ingestión de líquidos) y como agente aportador de energía.
El suero glucosado al 5 % proporciona, además, un aporte calórico nada despreciable. Cada litro de solución glucosada al 5 % aporta 50 gramos de glucosa, que equivale a 200 kcal. Este aporte calórico reduce el catabolismo proteico, y actúa por otra parte como protector hepático y como material de combustible de los tejidos del organismo más necesitados (sistema nervioso central y miocardio).
Las indicaciones principales de las soluciones isotónicas de glucosa al 5 % son la nutrición parenteral en enfermos con imposibilidad de aporte oral. Aquellos estados de deshidratación intracelular y extracelular como los que se producen en casos de vómitos, diarreas, fístulas intestinales, biliares y pancreáticas, estenosis pilórica, hemorragias, shock, sudación profusa, hiperventilación, poliurias, diabetes insípida, etc…, alteraciones del metabolismo hidrocarbonado que requieren de la administración de agua y glucosa.
Entre las contraindicaciones principales tenemos aquellas situaciones que puedan conducir a un cuadro grave de intoxicación acuosa por una sobrecarga desmesurada de solución glucosada, y enfermos addisonianos en los cuales se puede provocar una crisis addisoniana por edema celular e intoxicación acuosa.
Las soluciones de glucosa al 10 %, 20 % y 40 % son consideradas soluciones glucosadas hipertónicas, que al igual que la solución de glucosa isotónica, una vez metabolizadas desprenden energía y se transforma en agua. A su vez, y debido a que moviliza sodio desde la célula al espacio extracelular y potasio en sentido opuesto, se puede considerar a la glucosa como un proveedor indirecto de potasio a la célula.
La indicación más importante de las soluciones de glucosa hipertónica es el tratamiento del colapso circulatorio y de los edemas cerebral y pulmonar, porque la glucosa produciría una deshidratación celular y atraería agua hacia el espacio vascular, disminuyendo así la presión del líquido cefalorraquídeo y a nivel pulmonar.
Las soluciones glucosalinas (314 mOsm/L) son eficaces como hidratantes y para cubrir la demanda de agua y electrolitos. Cada litro de infusión de suero glucosalino aporta 35 gramos de glucosa (140 kcal), 60 mEq de sodio y 60 mEq de cloro.
Soluciones de uso en situaciones específicas.
Dentro de dichas soluciones de utilización en situaciones específicas, citaremos únicamente las de uso más habitual.
Estas soluciones se utilizan en aquellas situaciones que exista o se produzca una acidosis metabólica. El bicarbonato sódico fue el primer medicamento que se utilizó como tampón. El tamponamiento de un mmol de H+ conduce a la formación de un mmol de CO2, que debe ser eliminado por la vía respiratoria.
Para el uso clínico disponemos de varias presentaciones según las concentraciones a que se encuentren. Las de utilización más habitual son la solución de bicarbonato 1 Molar (1 M = 8.4 %), que sería la forma preferida para la corrección de la acidosis metabólica aguda, y la solución de bicarbonato 1/6 Molar (1.4 %) con osmolaridad semejante a la del plasma. La solución 1/6 Molar es la más empleada y su posología se realiza en función del déficit de bases y del peso del paciente.
El cloruro amónico 1/6 Molar es una solución isotónica (osmolaridad = 334), acidificante, de utilidad en el tratamiento de la alcalosis hipoclorémica.
El ión amonio es un dador de protones que se disocia en H+ y NH3+ , y su constante de disociación es tal que en la gama de pH de la sangre el NH4+ constituye el 99 % del amoníaco total. La acción acidificante depende de la conversión de los iones amonio en urea por el hígado, con generación de protones. Por ello, las soluciones de sales de amonio están contraindicadas en la insuficiencia hepática. Además, el cloruro de amonio posee toxicidad cuando es administrado de forma rápida, y puede desencadenar bradicardia, alteraciones respiratorias y contracciones musculares.
SOLUCIONES COLOIDALES.
Las soluciones coloidales contienen partículas en suspensión de alto peso molecular que no atraviesan las membranas capilares, de forma que son capaces de aumentar la presión osmótica plasmática y retener agua en el espacio intravascular. Así pues, las soluciones coloidales incrementan la presión oncótica y la efectividad del movimiento de fluidos desde el compartimento intersticial al compartimento plasmático deficiente. Es lo que se conoce como agente expansor plasmático. Producen efectos hemodinámicos más rápidos y sostenidos que las soluciones cristaloides, precisándose menos volumen que las soluciones cristaloides, aunque su coste es mayor.
Las características que debería poseer una solución coloidal son:
2.-Ausencia de otras acciones farmacológicas.
Podemos hacer una clasificación de los coloides como:
1) Soluciones coloidales naturales.
2) Soluciones coloidales artificiales.
Soluciones Coloidales Naturales.
Albumina.
La albúmina se produce en el hígado y es responsable de aproximadamente un 70-80 % de la presión oncótica del plasma, constituyendo un coloide efectivo. Su peso molecular oscila entre 66.300 y 66.900. La albúmina se distribuye entre los compartimentos intravascular (40 %) e intersticial (60 %). Su síntesis es estimulada por el cortisol y hormonas tiroideas, mientras que su producción disminuye cuando aumenta la presión oncótica del plasma. La concentración sérica normal en suero es de 3.5 a 5.0 g/dL y está correlacionado con el estado nutricional del sujeto. Si disminuyese la concentración de albúmina en el espacio intravascular, la albúmina del intersticio pasaría al espacio vascular a través de los canales linfáticos o bien por reflujo transcapilar.
La albúmina administrada se distribuye completamente dentro del espacio intravascular en dos minutos y tiene aproximadamente una vida media entre 4 y 16 horas. El 90 % de la albúmina administrada permanece en el plasma unas dos horas tras la administración, para posteriormente equilibrarse entre los espacios intra y extravascular durante un período de tiempo entre 7 a 10 días.Un 75 % de la albúmina comienza a desaparecer del plasma en 2 días. Su catabolismo tiene lugar en el tracto digestivo, riñones y sistema fagocítico mononuclear.
La albúmina es obtenida más comúnmente de plasma humano anticoagulado mediante el proceso de Cohn. En otros países, la placenta humana es utilizada como fuente para la obtención de albúmina.
Las soluciones de albúmina son esterilizadas mediante pasteurización a 60 ºC durante 10 horas, lo cual es efectivo para destruir los virus de la inmunodeficiencia humana, de las hepatitis B y no-A no-B (entre ellos el virus de la hepatitis C) 1. Sin embargo, pueden ser portadoras de pirógenos e infecciones bacterianas por contaminación de las soluciones. Incluso la pasteurización de la solución, puede provocar una polimerización de la albúmina creando una macromolécula con capacidad antigénica y de producir, por lo tanto, una reacción alérgica.
Las soluciones de albúmina contienen citrato, por lo que pueden ligarse al calcio sérico y derivar con ello una disminución de la función ventricular izquierda e incrementar el riesgo de insuficiencia renal. Por otra parte también pueden causar sangrado secundario a la disminución de la agregación plaquetaria y a una mayor dilución tanto de plaquetas como de los factores de la coagulación. Sin embargo, la albúmina causa menos cambios en los tiempos de protrombina, tiempo parcial de protrombina, y tiempo de coagulación.
Fracciones Proteicas de Plasma Humano.
Las fracciones proteicas del plasma, al igual que la albúmina, se obtiene por fraccionamientos seriados del plasma humano. La fracción proteica debe contener al menos 83 % de albúmina y no más de un 1 % de g-globulina, el resto estará formado por a y b-globulinas. Esta solución de fracciones proteicas está disponible como solución al 5 % en suero fisiológico y estabilizado con caprilato y acetiltrifosfanato sódico. Y al igual que la albúmina, estas soluciones son pasteurizadas a 60 ºC durante 10 horas.
Esta solución de fracciones proteicas tiene propiedades similares a la albúmina. La principal ventaja de esta solución consiste en su fácil manufacturación y la gran cantidad de proteínas aportadas.
Soluciones Coloidales Artificiales.
Dextranos.
Los dextranos son polisacáridos de origen bacteriano producidos por el Leuconostoc mesenteroides. Tiene propiedades oncóticas adecuadas pero no es capaz de transportar oxígeno. Mediante hidrólisis parcial y fraccionamiento de las largas moléculas nativas, el dextrán puede ser convertido en polisacáridos de cualquier peso molecular deseado.
La eliminación de los dextranos se realiza fundamentalmente por vía renal. La filtración glomerular de dextrano es dependiente del tamaño molecular. De este modo, podemos estimar que a las 6 horas de la administración del dextrano-40, alrededor del 60 % se ha eliminado por vía renal, frente a un 30 % de excreción del dextrano-70. A las 24 horas se habrá eliminado el 70 % del dextrano-40 y el 40 % del dextrano-70. Otra vía de eliminación es la digestiva por medio de las secreciones intestinales y pancreáticas ( 10 20 % de los dextranos ). Por último, una mínima parte es almacenada a nivel del hígado, bazo y riñones para ser degradada completamente a CO2 y H2O bajo la acción de una enzima específica, la dextrano 1-6 glucosidasa.
Otro de los posibles efectos indeseables de los dextranos sería la aparición de reacciones anafilácticas debidas a las IgG e IgM que pueden tener los dextranos. Algunos autores recomiendan la prevención de estas reacciones con una inyección previa, unos 15 mL, de dextrano de muy bajo peso molecular, que saturaría los sitios de fijación de las inmunoglobulinas, sin desencadenar una reacción inmunológica. No obstante, la incidencia de reacciones por hipersensibilidad ha disminuído en parte, porque las técnicas de preparación de las soluciones han sido mejoradas.
Hidroxietil-almidón (HEA).
El hetaalmidón es un almidón sintético, que se prepara a partir de amilopectina mediante la introducción de grupos hidroxietil éter en sus residuos de glucosa. El propósito de esta modificación es retardar la degradación del polímero por medio de las alfa-amilasas plasmáticas.
Dependiendo del grado de hidroxietilación y del peso molecular de las cadenas ramificadas de amilopectina será la duración de su efecto volémico, su metabolismo plasmático y la velocidad de eliminación renal. El hetaalmidón tiene un peso molecular promedio de 450.000, con límites entre 10.000 y 1.000.000. Las moléculas con peso molecular más bajo se excretan fácilmente por orina y, con el preparado habitual, alrededor del 40 % de la dosis es excretada en 24 horas 48. Las moléculas de peso molecular mayor son metabolizadas más lentamente; sólo alrededor del 1 % de la dosis persiste al cabo de dos semanas. Otra vía de eliminación del HEA es el tracto gastrointestinal y el sistema fagocítico mononuclear.
Está disponible para su uso clínico en soluciones al 6 % (60 gr/L) en solución salina isotónica al 0.9 %. Esta preparación es muy semejante a la del dextrán, y como él se emplea por sus propiedades oncóticas, pero se considera que el hetaalmidón es menos antigénico. La solución al 6 % tiene una presión oncótica de 30 mm Hg. La expansión aguda de volumen producida por el HEA es equivalente a la producida por la albúmina al 5 %, pero con una vida media sérica más prolongada, manteniendo un 50 % del efecto osmótico a las 24 horas.
Los efectos adversos del HEA son similares a los de otros coloides e incluyen las reacciónes alérgicas (aunque son menos frecuentes como indicamos anteriormente), precipitación de fallo cardíaco congestivo y fallo renal.
Pentaalmidón.
El pentaalmidón es un preparado con formulación semejante al hetaalmidón, pero con un peso molecular de 280.000 daltons y un número molecular medio de 120.000 daltons, por lo que también puede ser llamado hetaalmidón de bajo peso molecular. Se comercializa en solución al 10 %. El 90 % del producto es aclarado en unas 24 horas y prácticamente se hace indetectable a los 3 días. Su efecto expansor de volumen viene a durar unas 12 horas. Debido a su elevada presión oncótica, alrededor de 40 mm Hg, produce una de expansión de volumen superior a la que pudieran producir la albúmina al 5 % o el hetaalmidón al 6 %. Provoca un aumento de volumen de hasta 1.5 veces el volumen infundido.
Este producto actualmente no es aconsejado para utilizarlo como fluído de resucitación, únicamente es aprovechable en la leucoferesis. Entre sus posibles efectos adversos, se incluyen defectos de la coagulación secundarios a la hemodilución similares a los visto con el hetaalmidón, pero generalmente menos importantes.
Derivados de la gelatina.
Las soluciones de gelatina se emplearon por primera vez durante la 1ª Guerra Mundial, debido a su elevada viscosidad y bajo punto de congelación, y se han ido transformando hasta llegar a las gelatinas actuales.
Las gelatinas son polipéptidos obtenidos por desintegración del colágeno, y podemos distinguir 3 grupos:
1) Oxipoligelatinas
2) Gelatinas fluidas modificadas
3) Gelatinas modificadas con puentes de urea (estas dos últimas, las gelatinas fluidas y las modificadas con puentes de urea, se obtienen de colágeno bovino). La de utilización más frecuente es la modificada con puentes de urea, comúnmente conocida como Hemocé, que consiste en una solución de polipéptidos al 3.5 % obtenida después de de un proceso de disociación térmica y posterior polimerización reticular mediantes puentes de urea. Posee un peso molecular aproximado de 35.000 y una distribución entre 10.000 y 100.000. Estos polipéptidos están formados por 18 aminoácidos que suponen un aporte de nitrógeno de 6.3 gr/l de la solución al 3.5 %.Estas soluciones poseen un alto contenido en calcio (6 mmol/L) y en potasio (5 mmol/L), igualmente resulta ligeramente hiperoncótica.
Su eliminación es esencialmente renal. A las 4 horas de la administración los niveles séricos de gelatina modificada son ligeramente superiores al 40 % de la cantidad infundida. Transcurridas 12 horas, la cantidad que permanece aún en el espacio vascular es del 27 % y a las 48 horas se ha eliminada prácticamente toda. Esta capacidad de poder eliminarse tan fácilmente es lo que permite la utilización de elevadas cantidades de este coloide.
El efecto volumétrico se encuentra entre el 65 y el 70 % del volumen total administrado, disminuyendo progresivamente durante las 4 horas siguientes. Tiene una capacidad de retener agua en torno a 14 y 39 mL/g. A fin de obtener una reposición adecuada del volumen intravascular deben administrarse cantidades superiores a l déficit plasmático en un 30 %. Así pues, las características principales de este tipo de coloide son eliminación rápida, pero de efecto leve y corto.
